

Genomic characterization of SARS-CoV-2 from Uganda using MinION nanopore sequencing
- PMID: 37993530
- PMCID: PMC10665338
- DOI: 10.1038/s41598-023-47379-z
Genomic characterization of SARS-CoV-2 from Uganda using MinION nanopore sequencing
Abstract
SARS-CoV-2 undergoes frequent mutations, affecting COVID-19 diagnostics, transmission and vaccine efficacy. Here, we describe the genetic diversity of 49 SARS-CoV-2 samples from Uganda, collected during the COVID-19 waves of 2020/2021. Overall, the samples were similar to previously reported SARS-CoV-2 from Uganda and the Democratic Republic of Congo (DRC). The main lineages were AY.46 and A.23, which are considered to be Delta SARS-CoV-2 variants. Further, a total of 268 unique single nucleotide variants and 1456 mutations were found, with more than seventy percent mutations in the ORF1ab and S genes. The most common mutations were 2042C>G (83.4%), 14143C>T (79.5%), 245T>C (65%), and 1129G>T (51%), which occurred in the S, ORF1ab, ORF7a and N genes, respectively. As well, 28 structural variants-21 insertions and 7 deletions, occurred in 16 samples. Our findings point to the possibility that most SARS-CoV-2 infections in Uganda at the time arose from local spread and were not newly imported. Moreover, the relatedness of variants from Uganda and the DRC reflects high human mobility and interaction between the two countries, which is peculiar to this region of the world.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
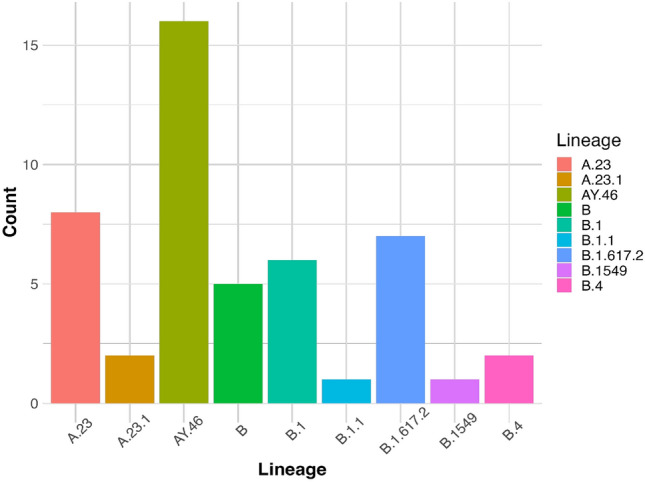

References
-
- World Health Organization . COVID-19 Weekly Epidemiological Update. World Health Organization; 2022. pp. 1–33.
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
