

Fast multiple-trait genome-wide association analysis for correlated longitudinal measurements
- PMID: 37996550
- PMCID: PMC10667366
- DOI: 10.1038/s41598-023-47555-1
Fast multiple-trait genome-wide association analysis for correlated longitudinal measurements
Abstract
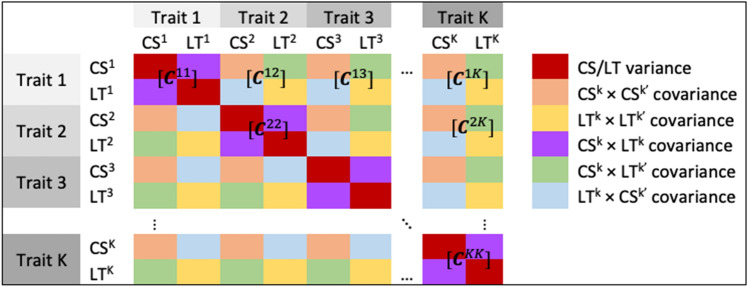
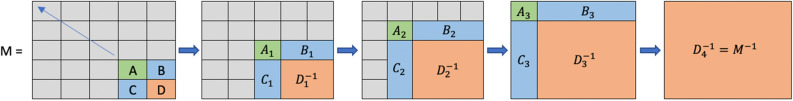
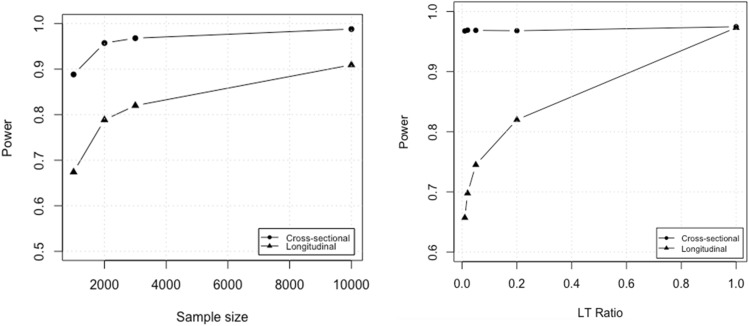
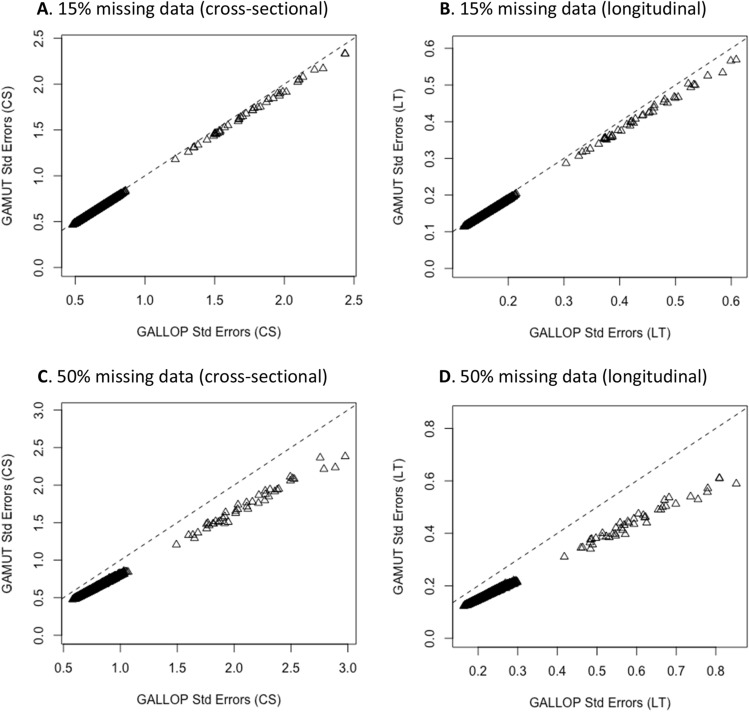
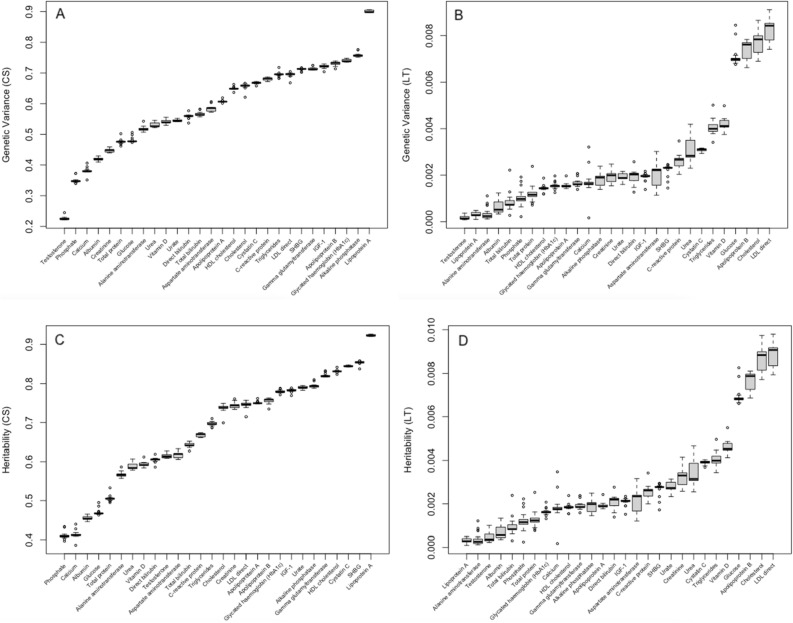
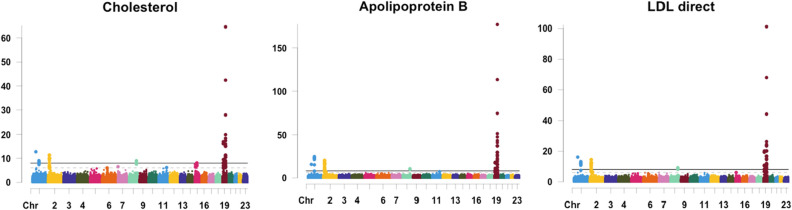
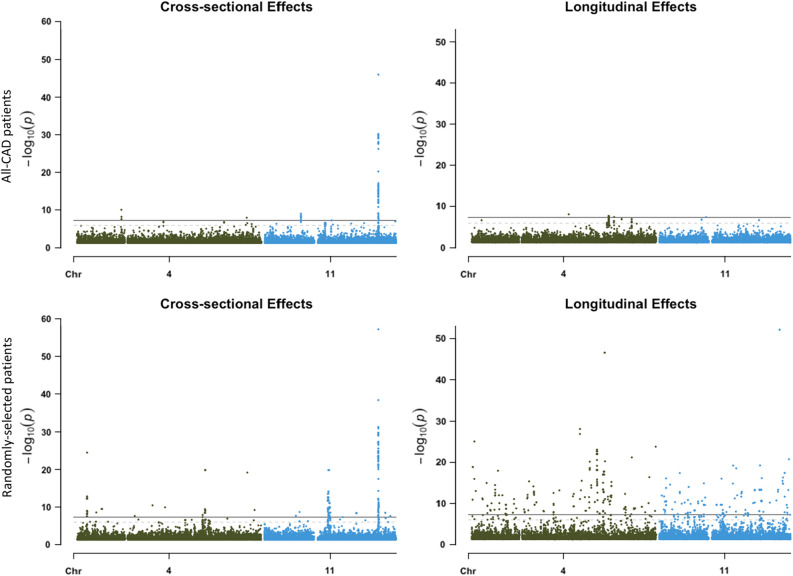
Large-scale longitudinal biobank data can be leveraged to identify genetic variation contributing to human diseases progression and traits trajectories. While methods for genome-wide association studies (GWAS) of multiple correlated traits have been proposed, an efficient multiple-trait approach to model longitudinal phenotypes is not currently available. We developed GAMUT, a genome-wide association approach for multiple longitudinal traits. GAMUT employs a mixed-effects model to fit longitudinal outcomes where a fast algorithm for inversion by recursive partitioning of the random effects submatrix is introduced. To evaluate performance of the algorithms introduced and assess their statistical power and type I error, stochastic simulation was conducted. Consistent with our expectation, power was greater for cross-sectional (CS) than longitudinal (LT) effects, particularly with a diminishing LT/CS ratio. With a minimum minor allele count of 3 within genotype by time categories, observed type I error was roughly equal to theoretical genome-wide significance. Additionally, 28 blood-based biomarkers measured at 2 time points on participants of the UK Biobank were used to compare GAMUT against single-trait standard and longitudinal GWAS (including rate of change). Across all biomarkers, we observed 539 (CS) and 248 (LT) significant independent variants for the GAMUT method, and 513 (CS) and 30 (LT) for single-trait longitudinal GWAS, respectively. Only 37 variants were identified by modeling rates of change using standard GWAS.
© 2023. The Author(s).
Conflict of interest statement
G. Abdel-Azim, L. Shuwei, and S. Guo are full-time employees of Johnson & Johnson. M. H. Black is a full-time employee of Foresite Labs. P. Patel is a full-time employee of Illumina.
Figures







References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
