

Accurate prediction of protein-nucleic acid complexes using RoseTTAFoldNA
- PMID: 37996753
- PMCID: PMC10776382
- DOI: 10.1038/s41592-023-02086-5
Accurate prediction of protein-nucleic acid complexes using RoseTTAFoldNA
Abstract
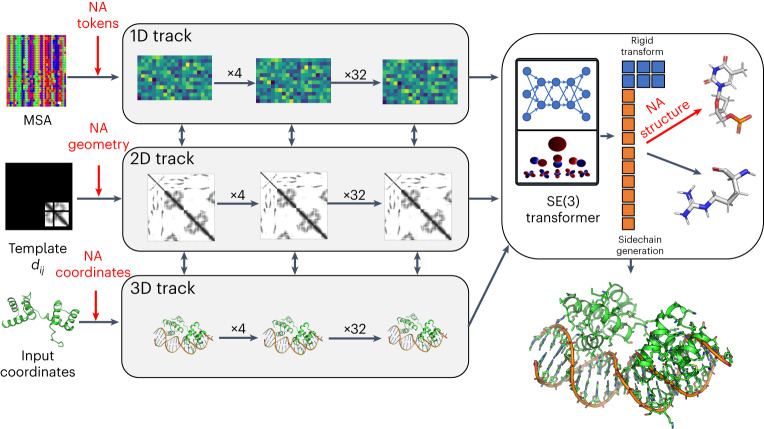
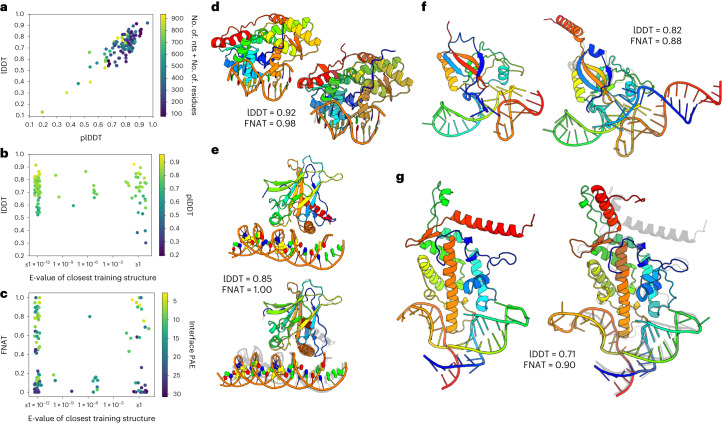
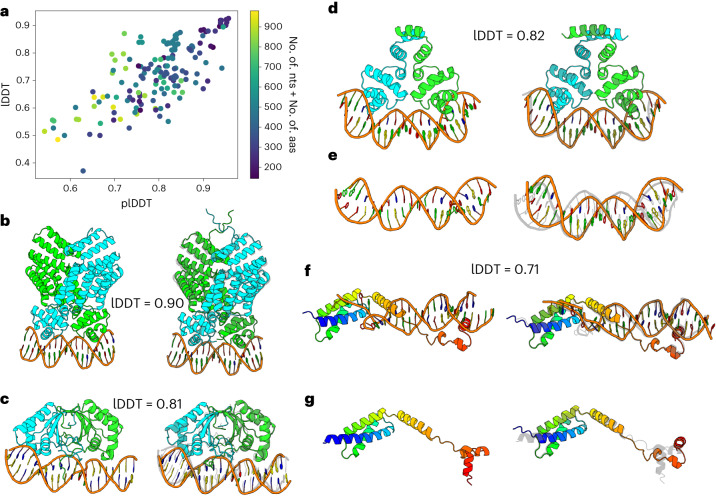
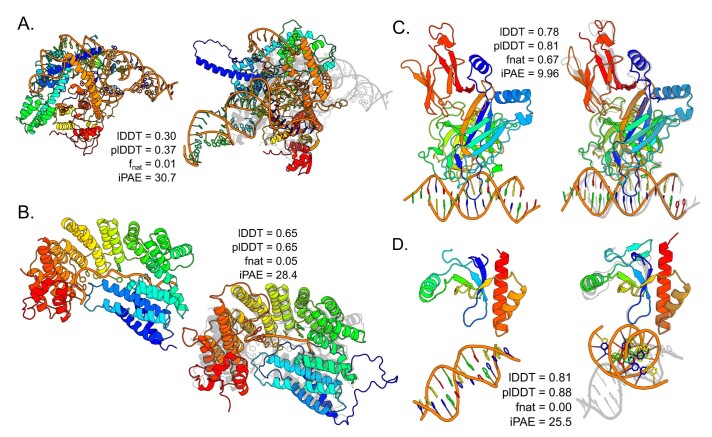
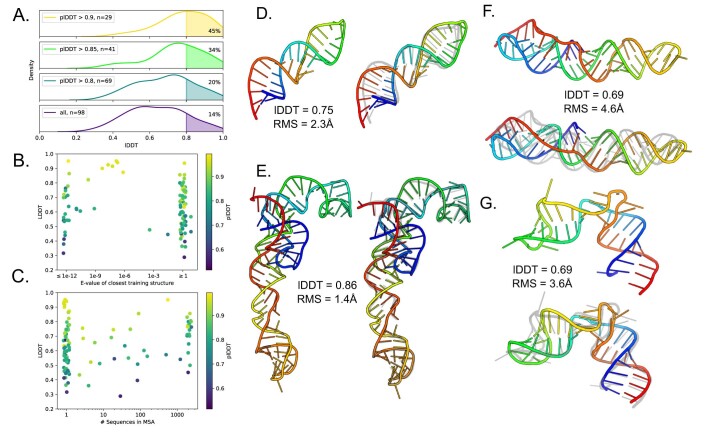
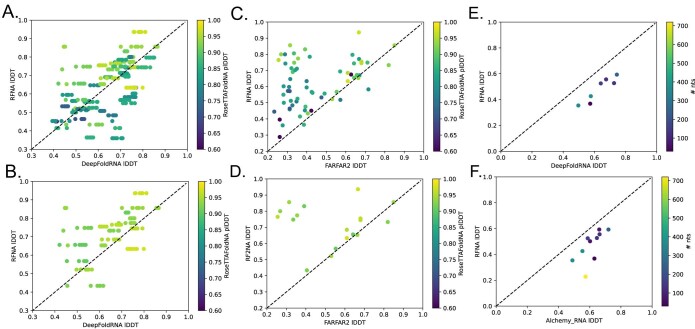
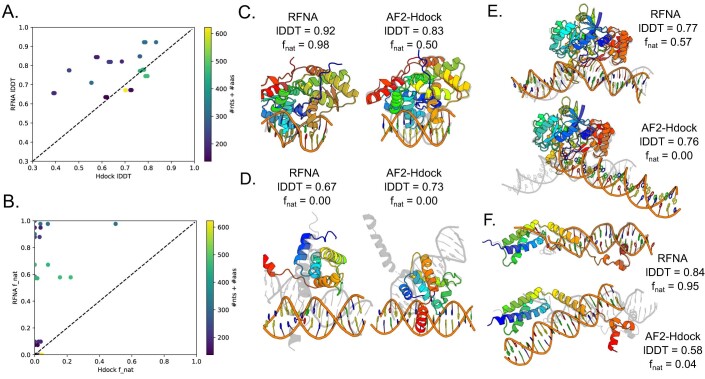
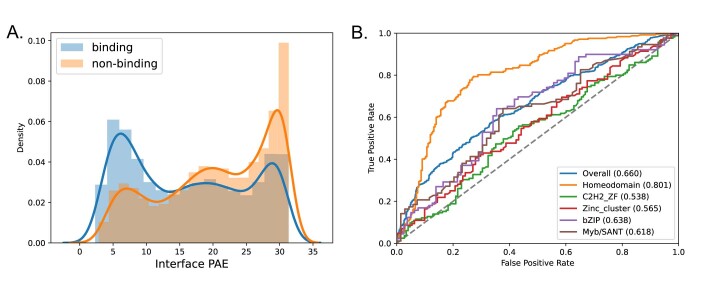
Protein-RNA and protein-DNA complexes play critical roles in biology. Despite considerable recent advances in protein structure prediction, the prediction of the structures of protein-nucleic acid complexes without homology to known complexes is a largely unsolved problem. Here we extend the RoseTTAFold machine learning protein-structure-prediction approach to additionally predict nucleic acid and protein-nucleic acid complexes. We develop a single trained network, RoseTTAFoldNA, that rapidly produces three-dimensional structure models with confidence estimates for protein-DNA and protein-RNA complexes. Here we show that confident predictions have considerably higher accuracy than current state-of-the-art methods. RoseTTAFoldNA should be broadly useful for modeling the structure of naturally occurring protein-nucleic acid complexes, and for designing sequence-specific RNA and DNA-binding proteins.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
