

CREaTor: zero-shot cis-regulatory pattern modeling with attention mechanisms
- PMID: 37996959
- PMCID: PMC10666311
- DOI: 10.1186/s13059-023-03103-8
CREaTor: zero-shot cis-regulatory pattern modeling with attention mechanisms
Abstract
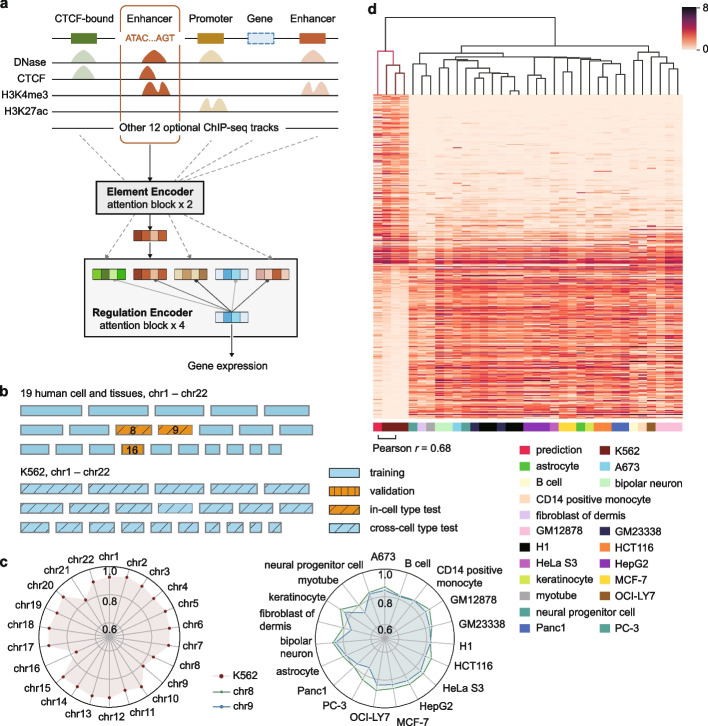
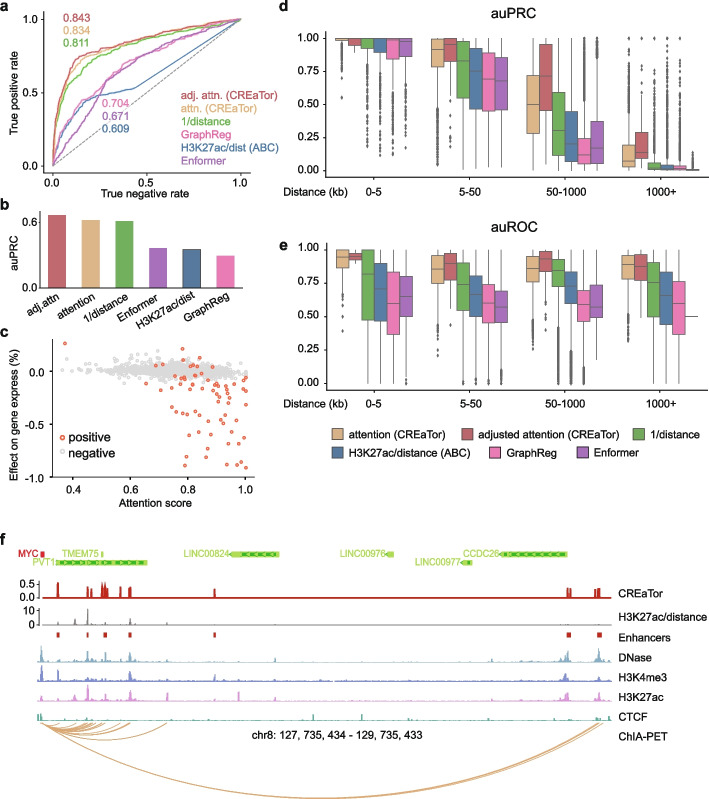
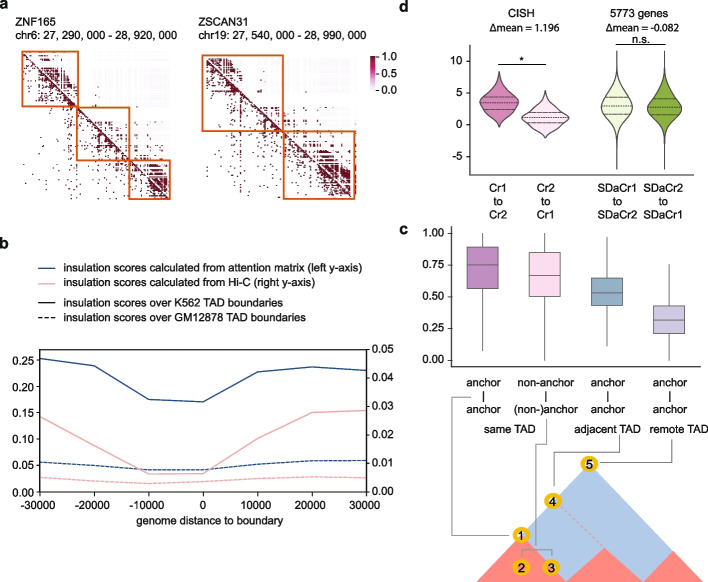
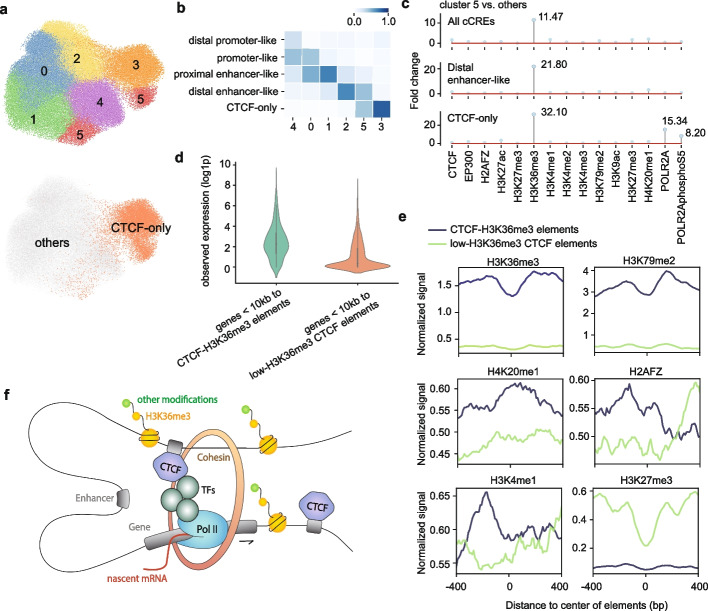
Linking cis-regulatory sequences to target genes has been a long-standing challenge. In this study, we introduce CREaTor, an attention-based deep neural network designed to model cis-regulatory patterns for genomic elements up to 2 Mb from target genes. Coupled with a training strategy that predicts gene expression from flanking candidate cis-regulatory elements (cCREs), CREaTor can model cell type-specific cis-regulatory patterns in new cell types without prior knowledge of cCRE-gene interactions or additional training. The zero-shot modeling capability, combined with the use of only RNA-seq and ChIP-seq data, allows for the ready generalization of CREaTor to a broad range of cell types.
Keywords: Cis-regulatory pattern; Enhancer-gene interaction; Epigenetics; Gene expression; Gene regulation.
© 2023. The Author(s).
Conflict of interest statement
P.D., F.J., H.X., L.H., L.W., J.Z., and B.S. are paid employees of Microsoft Research. Y.L., Z.C., and Y.Q. declare no competing interests.
Figures
Similar articles
-
Neuronal MAPT expression is mediated by long-range interactions with cis-regulatory elements.Am J Hum Genet. 2024 Feb 1;111(2):259-279. doi: 10.1016/j.ajhg.2023.12.015. Epub 2024 Jan 16. Am J Hum Genet. 2024. PMID: 38232730 Free PMC article.
-
MAPT expression is mediated by long-range interactions with cis-regulatory elements.bioRxiv [Preprint]. 2023 Apr 11:2023.03.07.531520. doi: 10.1101/2023.03.07.531520. bioRxiv. 2023. Update in: Am J Hum Genet. 2024 Feb 1;111(2):259-279. doi: 10.1016/j.ajhg.2023.12.015. PMID: 37090552 Free PMC article. Updated. Preprint.
-
Chromatin interaction-aware gene regulatory modeling with graph attention networks.Genome Res. 2022 May;32(5):930-944. doi: 10.1101/gr.275870.121. Epub 2022 Apr 8. Genome Res. 2022. PMID: 35396274 Free PMC article.
-
Favorable genomic environments for cis-regulatory evolution: A novel theoretical framework.Semin Cell Dev Biol. 2016 Sep;57:2-10. doi: 10.1016/j.semcdb.2015.12.003. Epub 2015 Dec 8. Semin Cell Dev Biol. 2016. PMID: 26673387 Review.
-
Genomic sources of regulatory variation in cis and in trans.Cell Mol Life Sci. 2005 Aug;62(16):1779-83. doi: 10.1007/s00018-005-5064-9. Cell Mol Life Sci. 2005. PMID: 15968467 Free PMC article. Review.
Cited by
-
Foundation model for comprehensive transcriptional regulation analysis.Natl Sci Rev. 2024 Oct 14;11(11):nwae355. doi: 10.1093/nsr/nwae355. eCollection 2024 Nov. Natl Sci Rev. 2024. PMID: 39555104 Free PMC article. No abstract available.
-
Machine and deep learning methods for predicting 3D genome organization.ArXiv [Preprint]. 2024 Mar 4:arXiv:2403.03231v1. ArXiv. 2024. Update in: Methods Mol Biol. 2025;2856:357-400. doi: 10.1007/978-1-0716-4136-1_22. PMID: 38495565 Free PMC article. Updated. Preprint.
-
Machine and Deep Learning Methods for Predicting 3D Genome Organization.Methods Mol Biol. 2025;2856:357-400. doi: 10.1007/978-1-0716-4136-1_22. Methods Mol Biol. 2025. PMID: 39283464 Review.
References
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
