

From Stress to Sick(le) and Back Again-Oxidative/Antioxidant Mechanisms, Genetic Modulation, and Cerebrovascular Disease in Children with Sickle Cell Anemia
- PMID: 38001830
- PMCID: PMC10669666
- DOI: 10.3390/antiox12111977
From Stress to Sick(le) and Back Again-Oxidative/Antioxidant Mechanisms, Genetic Modulation, and Cerebrovascular Disease in Children with Sickle Cell Anemia
Abstract
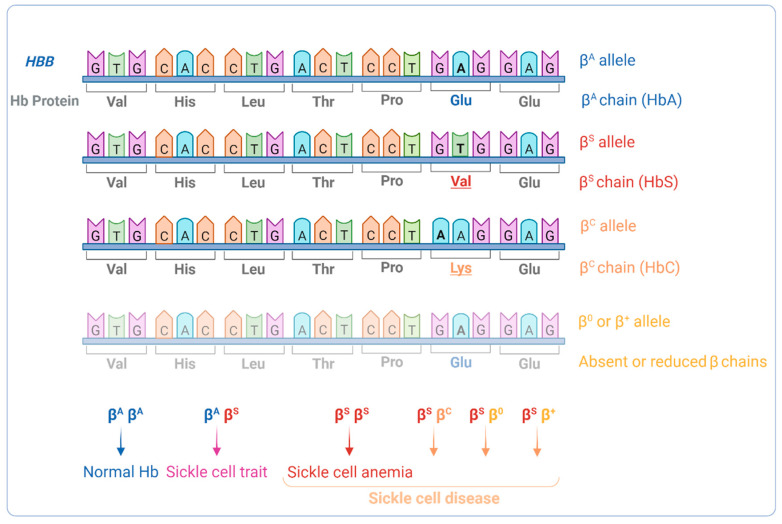
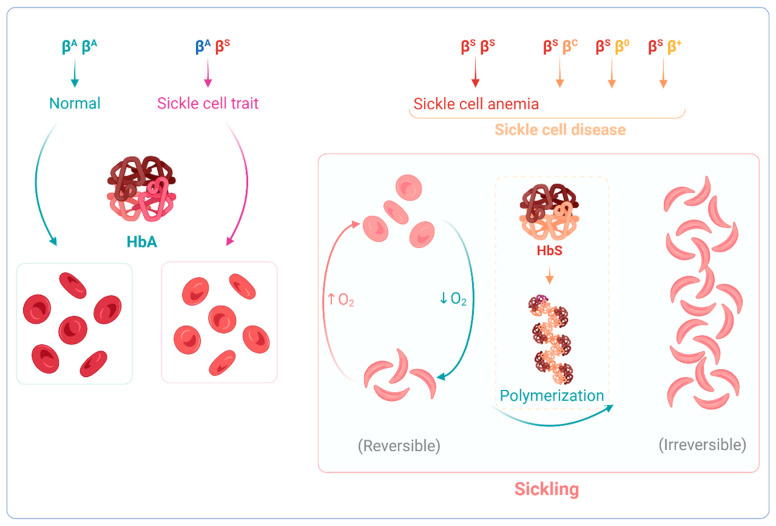
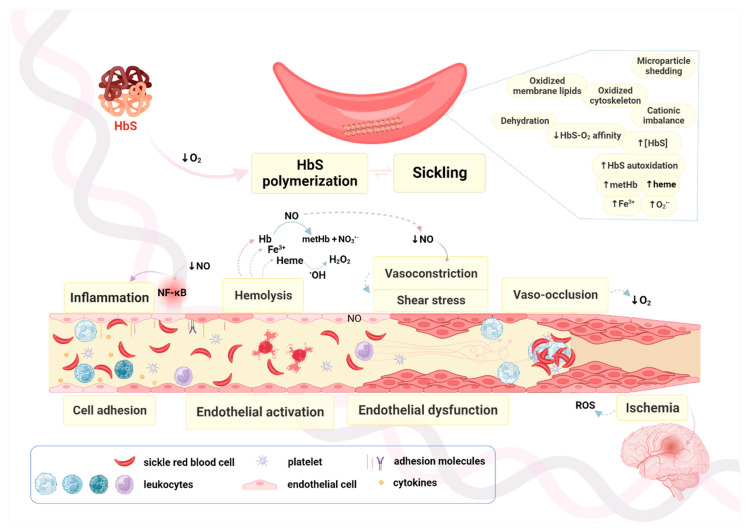
Sickle cell anemia (SCA) is a genetic disease caused by the homozygosity of the HBB:c.20A>T mutation, which results in the production of hemoglobin S (HbS). In hypoxic conditions, HbS suffers autoxidation and polymerizes inside red blood cells, altering their morphology into a sickle shape, with increased rigidity and fragility. This triggers complex pathophysiological mechanisms, including inflammation, cell adhesion, oxidative stress, and vaso-occlusion, along with metabolic alterations and endocrine complications. SCA is phenotypically heterogeneous due to the modulation of both environmental and genetic factors. Pediatric cerebrovascular disease (CVD), namely ischemic stroke and silent cerebral infarctions, is one of the most impactful manifestations. In this review, we highlight the role of oxidative stress in the pathophysiology of pediatric CVD. Since oxidative stress is an interdependent mechanism in vasculopathy, occurring alongside (or as result of) endothelial dysfunction, cell adhesion, inflammation, chronic hemolysis, ischemia-reperfusion injury, and vaso-occlusion, a brief overview of the main mechanisms involved is included. Moreover, the genetic modulation of CVD in SCA is discussed. The knowledge of the intricate network of altered mechanisms in SCA, and how it is affected by different genetic factors, is fundamental for the identification of potential therapeutic targets, drug development, and patient-specific treatment alternatives.
Keywords: antioxidant mechanisms; cerebrovascular disease; genetic modulators; oxidative stress; sickle cell anemia; vasculopathy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Hemorheological Alterations and Oxidative Damage in Sickle Cell Anemia.Front Mol Biosci. 2019 Dec 4;6:142. doi: 10.3389/fmolb.2019.00142. eCollection 2019. Front Mol Biosci. 2019. PMID: 31867341 Free PMC article.
-
Hemorheological alterations in sickle cell anemia and their clinical consequences - The role of genetic modulators.Clin Hemorheol Microcirc. 2016;64(4):859-866. doi: 10.3233/CH-168048. Clin Hemorheol Microcirc. 2016. PMID: 27814292
-
The Red Blood Cell-Inflammation Vicious Circle in Sickle Cell Disease.Front Immunol. 2020 Mar 13;11:454. doi: 10.3389/fimmu.2020.00454. eCollection 2020. Front Immunol. 2020. PMID: 32231672 Free PMC article. Review.
-
Sickle cell disease, vasculopathy, and therapeutics.Annu Rev Med. 2013;64:451-66. doi: 10.1146/annurev-med-120611-143127. Epub 2012 Nov 26. Annu Rev Med. 2013. PMID: 23190149 Review.
-
Genetic modulation of anemia severity, hemolysis level, and hospitalization rate in Angolan children with Sickle Cell Anemia.Mol Biol Rep. 2022 Nov;49(11):10347-10356. doi: 10.1007/s11033-022-07831-1. Epub 2022 Sep 12. Mol Biol Rep. 2022. PMID: 36097125
Cited by
-
Vitamin D Status and Oxidative Stress in Children with Sickle Cell Anaemia in Sagamu, Nigeria.Sultan Qaboos Univ Med J. 2025 May 2;25(1):105-113. doi: 10.18295/squmj.10.2024.054. eCollection 2025. Sultan Qaboos Univ Med J. 2025. PMID: 40657454 Free PMC article.
-
Crosstalk Between Sickle Cell Disease and Ferroptosis.Int J Mol Sci. 2025 Apr 13;26(8):3675. doi: 10.3390/ijms26083675. Int J Mol Sci. 2025. PMID: 40332185 Free PMC article. Review.
References
-
- Piel F.B., Tatem A.J., Huang Z., Gupta S., Williams T.N., Weatherall D.J. Global Migration and the Changing Distribution of Sickle Haemoglobin: A Quantitative Study of Temporal Trends between 1960 and 2000. Lancet Glob. Health. 2014;2:e80–e89. doi: 10.1016/S2214-109X(13)70150-5. - DOI - PMC - PubMed
-
- Debaun M.R., Kirkham F.J. Central Nervous System Complications and Management in Sickle Cell Disease. Blood. 2016;127:829–838. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
