

Homozygous Duplication in the CHRNE in a Family with Congenital Myasthenic Syndrome 4C: 18-Year Follow Up
- PMID: 38001983
- PMCID: PMC10668953
- DOI: 10.3390/biomedicines11112983
Homozygous Duplication in the CHRNE in a Family with Congenital Myasthenic Syndrome 4C: 18-Year Follow Up
Abstract
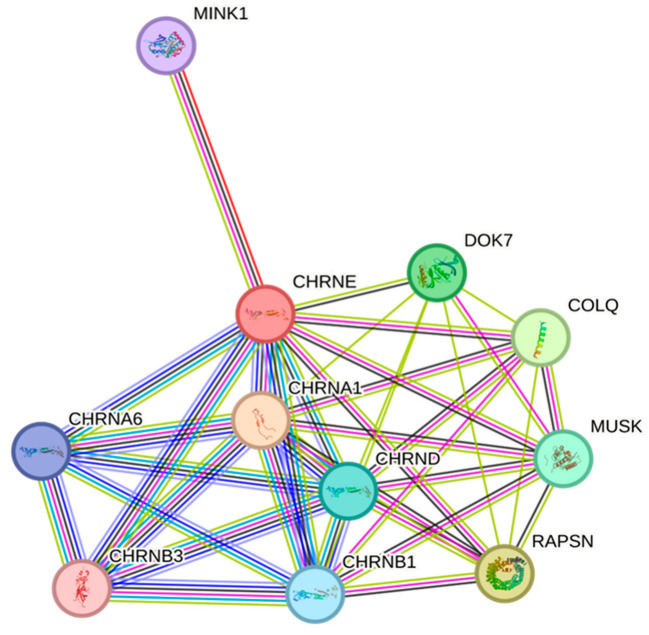
Background and objectives: Congenital myasthenic syndromes (CMSs) are rare inherited diseases characterized by muscle weakness and fatigability on exertion resulting from defects in the neuromuscular junctions. Mutations in 32 genes have been reported as the underlying causes of CMS, with mutations in the cholinergic receptor nicotinic epsilon subunit (CHRNE) being the most common cause of the disease. Methodology and Materials: This study investigated a large consanguineous family with multiple individuals suffering from abnormal fatigue and muscle weakness in the ocular and limb regions. Moreover, the affected individuals were followed up for 18 years to observe the clinical course of the disease.
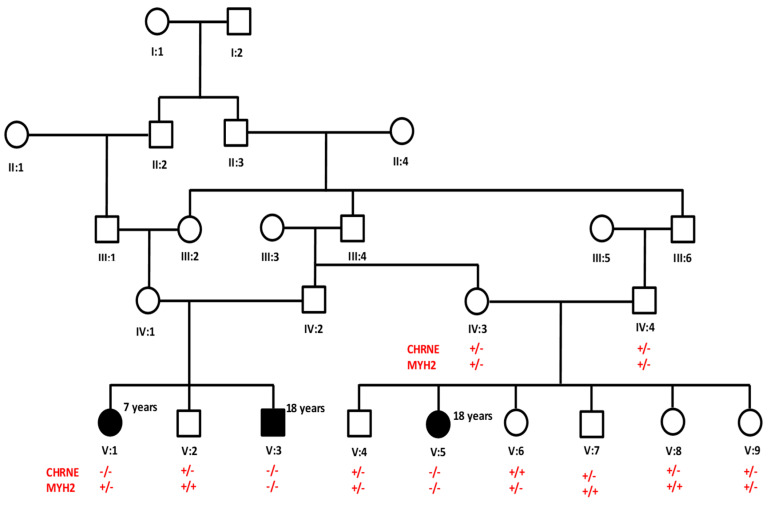
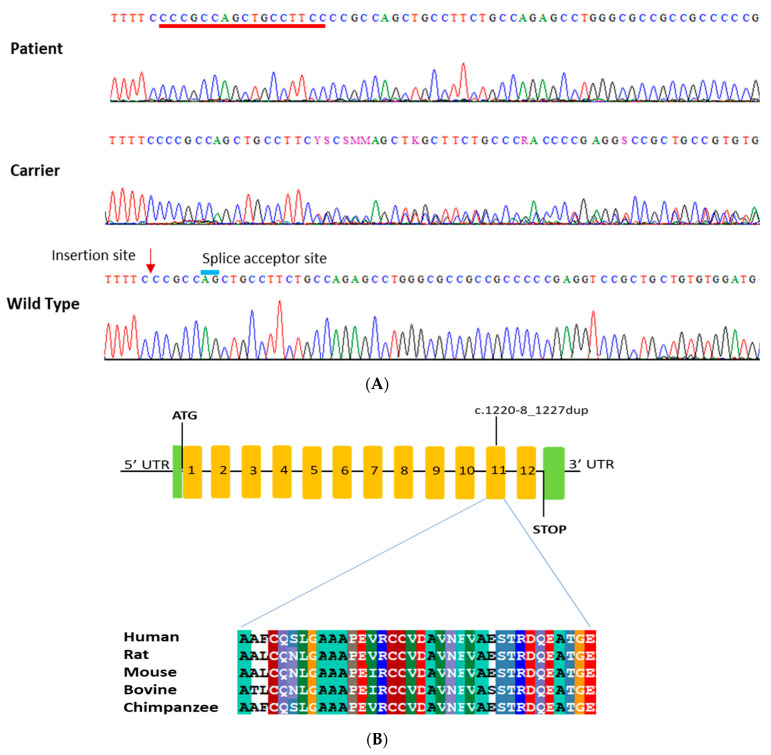
Results: High-quality exome sequencing followed by bidirectional Sanger sequencing revealed a homozygous duplication variant (NM_000080.4: c.1220-8_1227dup) in the splice acceptor site of exon 11 of the CHRNE gene. This variant is predicted to cause frameshift and premature termination (p.Cys410ProfsTer51). Both parents had heterozygous duplication variants with no clinical symptoms. The personalized treatment of the affected individuals resulted in a marked improvement in the clinical symptoms. More than 80% of the disease symptoms in the affected individuals subsided after the use of pyridostigmine and salbutamol (4 mg).
Conclusions: This is the first report of long-term follow up of cases with homozygous insertion (c.1220-8_1227dup) in the CHRNE gene. Furthermore, this report expands the phenotypic symptoms associated with the CHRNE mutation.
Keywords: CHRNE mutation; congenital myasthenic syndrome; genetics; phenotypic spectrum.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Characterization of Clinical Phenotypes in Congenital Myasthenic Syndrome Associated with the c.1327delG Frameshift Mutation in CHRNE Encoding the Acetylcholine Receptor Epsilon Subunit.J Neuromuscul Dis. 2024;11(5):1011-1020. doi: 10.3233/JND-230235. J Neuromuscul Dis. 2024. PMID: 38995797 Free PMC article.
-
A Novel Homozygous Variant in the CHRNE Gene in 2 Siblings with Congenital Myasthenic Syndrome.Child Neurol Open. 2023 Nov 28;10:2329048X231216432. doi: 10.1177/2329048X231216432. eCollection 2023 Jan-Dec. Child Neurol Open. 2023. PMID: 38034490 Free PMC article.
-
A common CHRNE mutation in Brazilian patients with congenital myasthenic syndrome.J Neurol. 2018 Mar;265(3):708-713. doi: 10.1007/s00415-018-8736-8. Epub 2018 Jan 30. J Neurol. 2018. PMID: 29383513 Free PMC article.
-
Congenital Myasthenic Syndrome associated with acetylcholine receptor deficiency: case report and review of the literature.Ophthalmic Genet. 2024 Oct;45(5):481-487. doi: 10.1080/13816810.2024.2352391. Epub 2024 Jun 4. Ophthalmic Genet. 2024. PMID: 38832364 Review.
-
Delineation of molecular characteristics of congenital myasthenic syndromes in Indian families and review of literature.Clin Dysmorphol. 2023 Oct 1;32(4):162-167. doi: 10.1097/MCD.0000000000000465. Epub 2023 Jun 19. Clin Dysmorphol. 2023. PMID: 37646703 Review.
Cited by
-
Exome Sequence Analysis to Characterize Undiagnosed Family Segregating Motor Impairment and Dystonia.J Clin Med. 2024 Jul 21;13(14):4252. doi: 10.3390/jcm13144252. J Clin Med. 2024. PMID: 39064292 Free PMC article.
-
Exome sequence analysis identifies a homozygous, pathogenic, frameshift variant in the MAN2B1 gene underlying clinical variant of α-mannosidosis.Front Genet. 2024 Aug 30;15:1421943. doi: 10.3389/fgene.2024.1421943. eCollection 2024. Front Genet. 2024. PMID: 39280098 Free PMC article.
-
Congenital Myasthenic Syndrome-4C in a Consanguineous Romani Family: Genetic Insights and Clinical Implications.Diagnostics (Basel). 2025 Jan 21;15(3):235. doi: 10.3390/diagnostics15030235. Diagnostics (Basel). 2025. PMID: 39941166 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
