

Exploiting TLK1 and Cisplatin Synergy for Synthetic Lethality in Androgen-Insensitive Prostate Cancer
- PMID: 38001987
- PMCID: PMC10669050
- DOI: 10.3390/biomedicines11112987
Exploiting TLK1 and Cisplatin Synergy for Synthetic Lethality in Androgen-Insensitive Prostate Cancer
Abstract
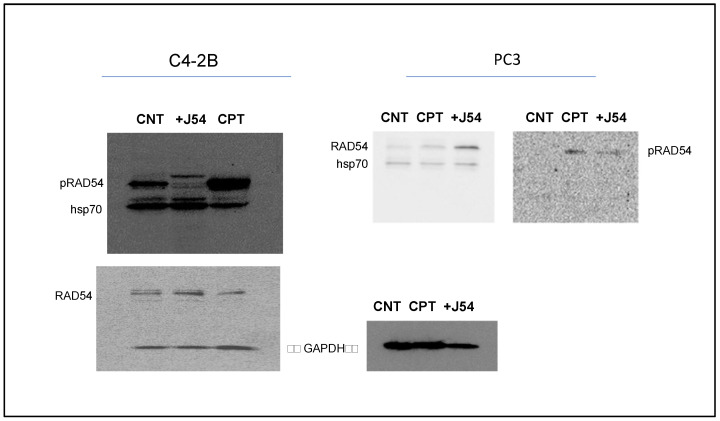
Cellular organisms possess intricate DNA damage repair and tolerance pathways to manage various DNA lesions arising from endogenous or exogenous sources. The dysregulation of these pathways is associated with cancer development and progression. Synthetic lethality (SL), a promising cancer therapy concept, involves exploiting the simultaneous functional loss of two genes for selective cell death. PARP inhibitors (PARPis) have demonstrated success in BRCA-deficient tumors. Cisplatin (CPT), a widely used chemotherapy agent, forms DNA adducts and crosslinks, rendering it effective against various cancers, but less so for prostate cancer (PCa) due to resistance and toxicity. Here, we explore the therapeutic potential of TLK1, a kinase upregulated in androgen-insensitive PCa cells, as a target for enhancing CPT-based therapy. TLK1 phosphorylates key homologous recombination repair (HRR) proteins, RAD54L and RAD54B, which are critical for HRR alongside RAD51. The combination of CPT with TLK1 inhibitor J54 exhibits SL in androgen-insensitive PCa cells. The formation of double-strand break intermediates during inter-strand crosslink processing necessitates HRR for effective repair. Therefore, targeting TLK1 with J54 enhances the SL of CPT by impeding HRR, leading to increased sensitivity in PCa cells. These findings suggest a promising approach for improving CPT-based therapies in PCa, particularly in androgen-insensitive cases. By elucidating the role of TLK1 in CPT resistance, this study provides valuable insights into potential therapeutic targets to overcome PCa resistance to CPT chemotherapy. Further investigations into TLK1 inhibition in combination with other DNA-damaging agents may pave the way for more effective and targeted treatments for PCa and other cancers that exhibit resistance to traditional chemotherapy agents.
Keywords: CPT-based PCa therapy; PCa; TLK1 inhibitor J54; TLK1 signaling; homologous recombination repair; synthetic lethality.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





Similar articles
-
Targeting Prostate Cancer, the 'Tousled Way'.Int J Mol Sci. 2023 Jul 5;24(13):11100. doi: 10.3390/ijms241311100. Int J Mol Sci. 2023. PMID: 37446279 Free PMC article. Review.
-
Interaction of TLK1 and AKTIP as a Potential Regulator of AKT Activation in Castration-Resistant Prostate Cancer Progression.Pathophysiology. 2021 Jul 20;28(3):339-354. doi: 10.3390/pathophysiology28030023. Pathophysiology. 2021. PMID: 35366279 Free PMC article.
-
The TLK1-MK5 Axis Regulates Motility, Invasion, and Metastasis of Prostate Cancer Cells.Cancers (Basel). 2022 Nov 22;14(23):5728. doi: 10.3390/cancers14235728. Cancers (Basel). 2022. PMID: 36497211 Free PMC article.
-
TLK1-mediated MK5-S354 phosphorylation drives prostate cancer cell motility and may signify distinct pathologies.Mol Oncol. 2022 Jul;16(13):2537-2557. doi: 10.1002/1878-0261.13183. Epub 2022 Feb 3. Mol Oncol. 2022. PMID: 35064619 Free PMC article.
-
DNA-Damage-Repair Gene Alterations in Genitourinary Malignancies.Eur Surg Res. 2022;63(4):155-164. doi: 10.1159/000526415. Epub 2022 Aug 9. Eur Surg Res. 2022. PMID: 35944490 Review.
Cited by
-
TLK1>Nek1 Axis Promotes Nuclear Retention and Activation of YAP with Implications for Castration-Resistant Prostate Cancer.Cancers (Basel). 2024 Aug 22;16(16):2918. doi: 10.3390/cancers16162918. Cancers (Basel). 2024. PMID: 39199688 Free PMC article.
-
Beyond the Horizon: Rethinking Prostate Cancer Treatment Through Innovation and Alternative Strategies.Cancers (Basel). 2024 Dec 29;17(1):75. doi: 10.3390/cancers17010075. Cancers (Basel). 2024. PMID: 39796704 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
