

Assessing Genetic Algorithm-Based Docking Protocols for Prediction of Heparin Oligosaccharide Binding Geometries onto Proteins
- PMID: 38002315
- PMCID: PMC10669598
- DOI: 10.3390/biom13111633
Assessing Genetic Algorithm-Based Docking Protocols for Prediction of Heparin Oligosaccharide Binding Geometries onto Proteins
Abstract
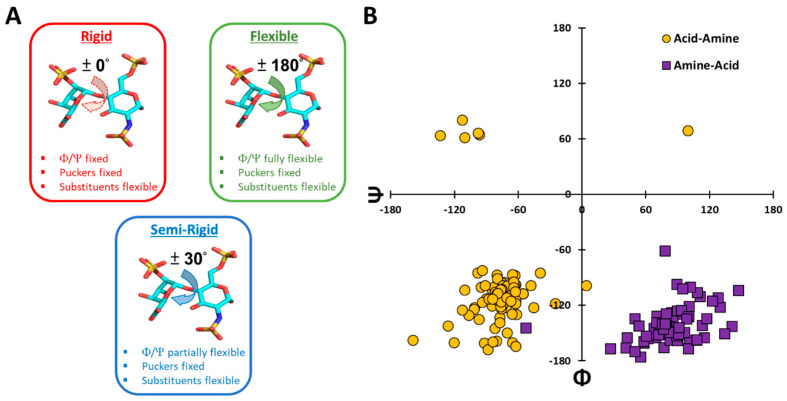
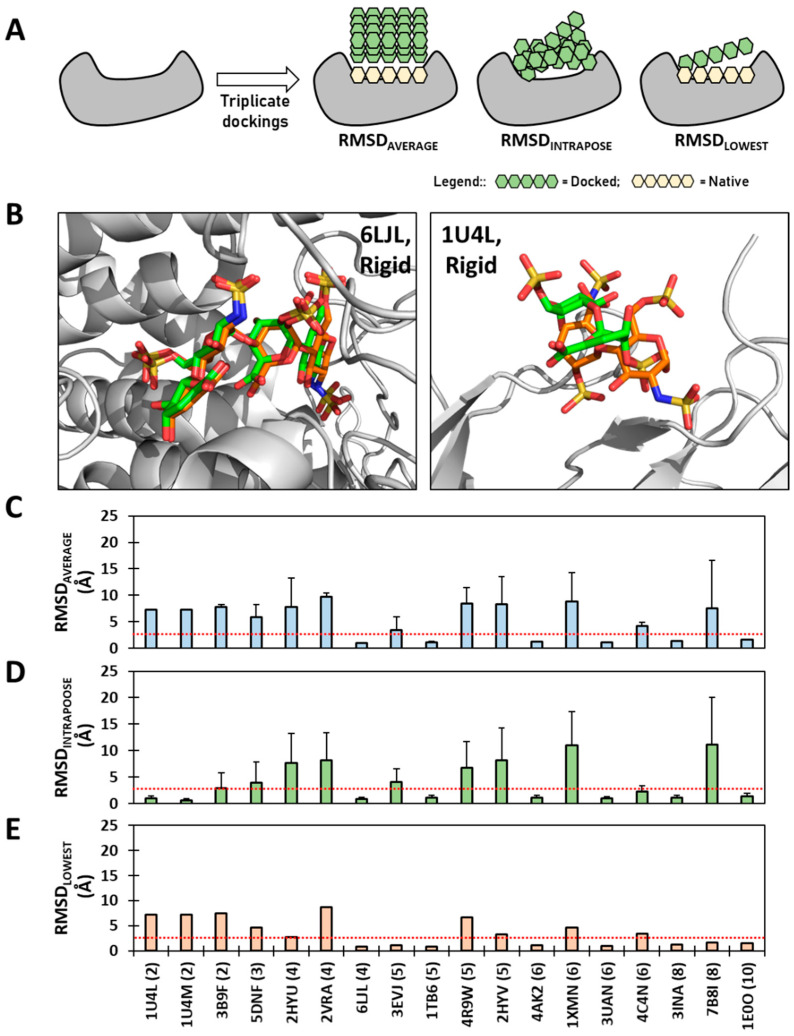
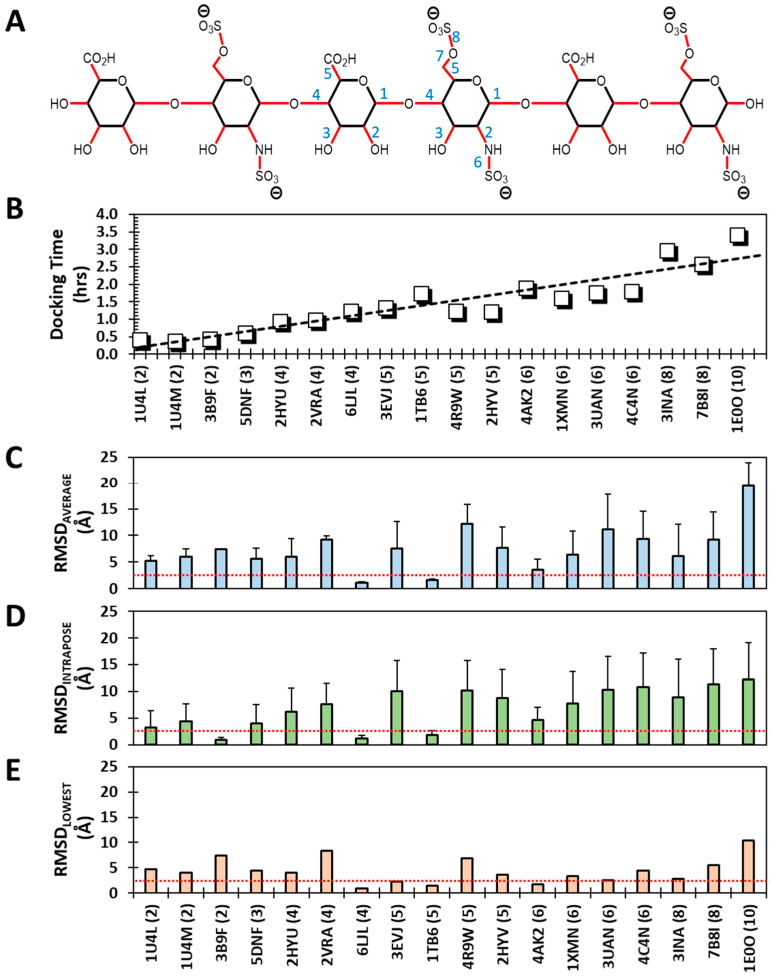
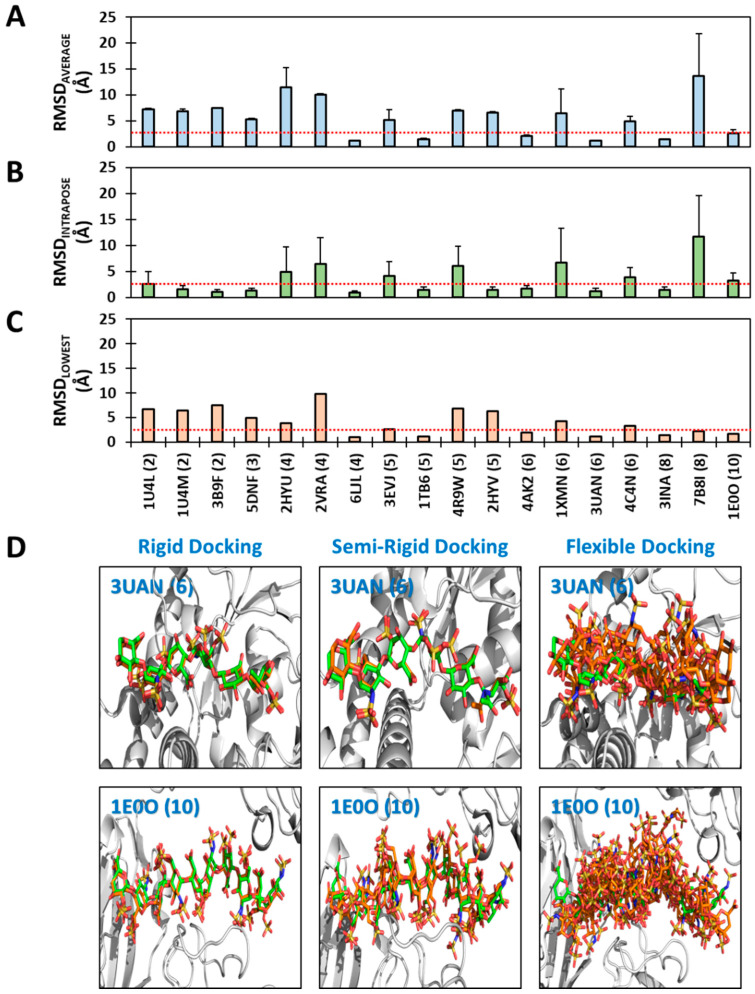
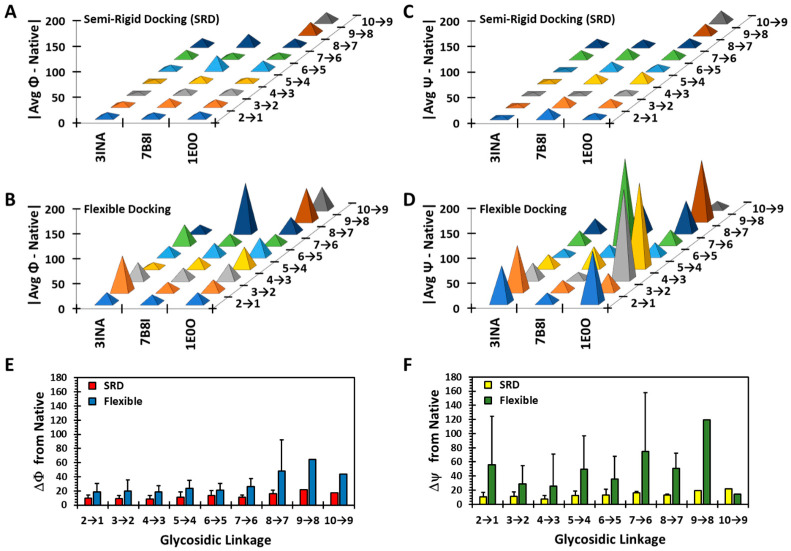
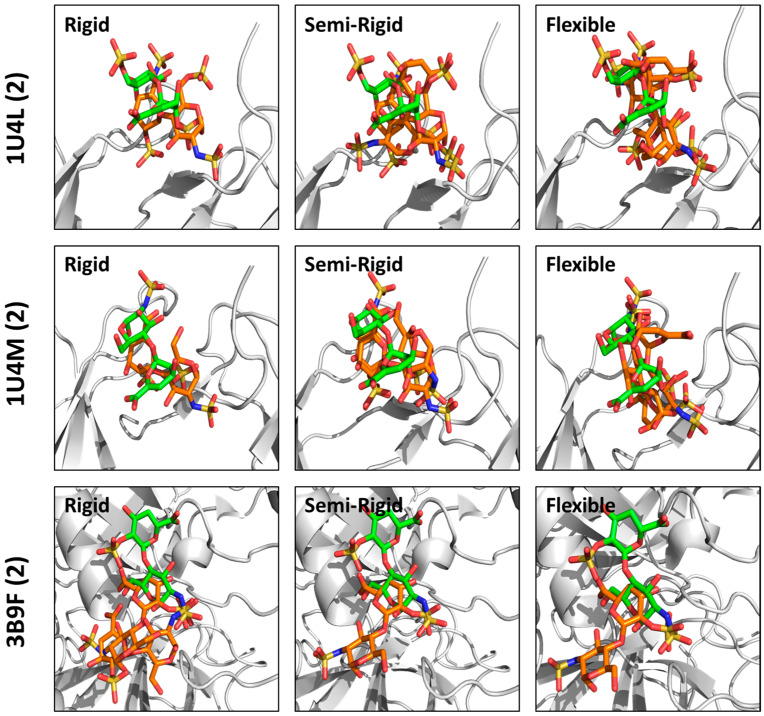
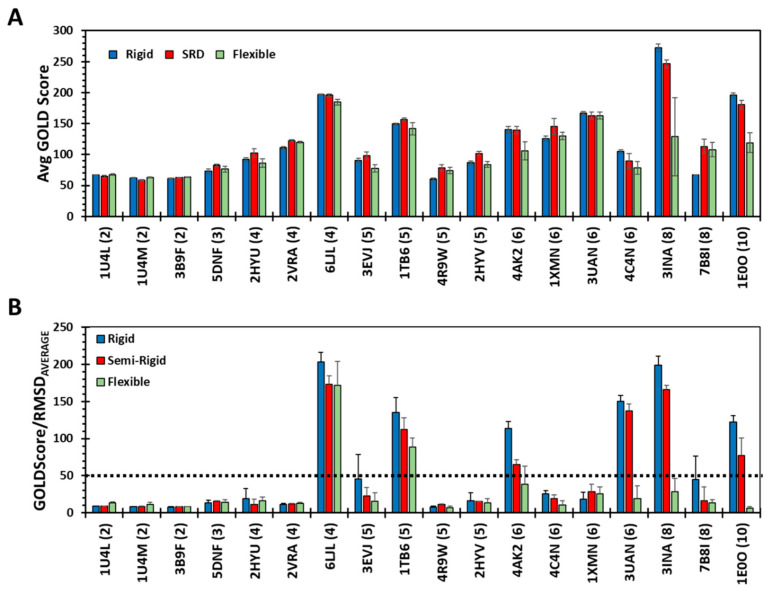
Although molecular docking has evolved dramatically over the years, its application to glycosaminoglycans (GAGs) has remained challenging because of their intrinsic flexibility, highly anionic character and rather ill-defined site of binding on proteins. GAGs have been treated as either fully "rigid" or fully "flexible" in molecular docking. We reasoned that an intermediate semi-rigid docking (SRD) protocol may be better for the recapitulation of native heparin/heparan sulfate (Hp/HS) topologies. Herein, we study 18 Hp/HS-protein co-complexes containing chains from disaccharide to decasaccharide using genetic algorithm-based docking with rigid, semi-rigid, and flexible docking protocols. Our work reveals that rigid and semi-rigid protocols recapitulate native poses for longer chains (5→10 mers) significantly better than the flexible protocol, while 2→4-mer poses are better predicted using the semi-rigid approach. More importantly, the semi-rigid docking protocol is likely to perform better when no crystal structure information is available. We also present a new parameter for parsing selective versus non-selective GAG-protein systems, which relies on two computational parameters including consistency of binding (i.e., RMSD) and docking score (i.e., GOLD Score). The new semi-rigid protocol in combination with the new computational parameter is expected to be particularly useful in high-throughput screening of GAG sequences for identifying promising druggable targets as well as drug-like Hp/HS sequences.
Keywords: glycosaminoglycans; heparin/heparan sulfate; knowledge-based docking; molecular docking.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Structural Insights into Endostatin-Heparan Sulfate Interactions Using Modeling Approaches.Molecules. 2024 Aug 26;29(17):4040. doi: 10.3390/molecules29174040. Molecules. 2024. PMID: 39274888 Free PMC article.
-
Mapping of heparin/heparan sulfate binding sites on αvβ3 integrin by molecular docking.J Mol Recognit. 2013 Feb;26(2):76-85. doi: 10.1002/jmr.2250. J Mol Recognit. 2013. PMID: 23334915
-
Docking glycosaminoglycans to proteins: analysis of solvent inclusion.J Comput Aided Mol Des. 2011 May;25(5):477-89. doi: 10.1007/s10822-011-9433-1. Epub 2011 May 20. J Comput Aided Mol Des. 2011. PMID: 21597992 Free PMC article.
-
Conformation and dynamics of heparin and heparan sulfate.Glycobiology. 2000 Nov;10(11):1147-56. doi: 10.1093/glycob/10.11.1147. Glycobiology. 2000. PMID: 11087707 Review.
-
Expedient Synthesis of Core Disaccharide Building Blocks from Natural Polysaccharides for Heparan Sulfate Oligosaccharide Assembly.Angew Chem Int Ed Engl. 2019 Dec 16;58(51):18577-18583. doi: 10.1002/anie.201908805. Epub 2019 Nov 6. Angew Chem Int Ed Engl. 2019. PMID: 31553820 Free PMC article. Review.
Cited by
-
The role of global DNA methylation in citrinin induced toxicity: In vitro and in silico approach.Toxicol Rep. 2025 May 9;14:102046. doi: 10.1016/j.toxrep.2025.102046. eCollection 2025 Jun. Toxicol Rep. 2025. PMID: 40487379 Free PMC article.
-
The chemical composition analysis of Yixin Tongmai Granules using UHPLC-MS/MS and exploration of its potential mechanism in treatment of coronary artery disease based on network pharmacology and molecular docking.Medicine (Baltimore). 2025 Feb 21;104(8):e41620. doi: 10.1097/MD.0000000000041620. Medicine (Baltimore). 2025. PMID: 39993113 Free PMC article.
-
Identification of an Unnatural Sulfated Monosaccharide as a High-Affinity Ligand for Pan-Variant Targeting of SARS-CoV-2 Spike Glycoprotein.ACS Chem Biol. 2025 Jun 20;20(6):1394-1405. doi: 10.1021/acschembio.5c00206. Epub 2025 May 13. ACS Chem Biol. 2025. PMID: 40358361 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
