

Animal Models for the Study of Gaucher Disease
- PMID: 38003227
- PMCID: PMC10671165
- DOI: 10.3390/ijms242216035
Animal Models for the Study of Gaucher Disease
Abstract
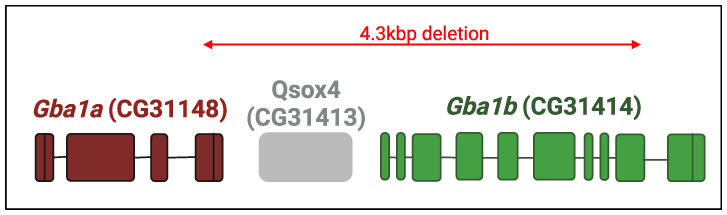
In Gaucher disease (GD), a relatively common sphingolipidosis, the mutant lysosomal enzyme acid β-glucocerebrosidase (GCase), encoded by the GBA1 gene, fails to properly hydrolyze the sphingolipid glucosylceramide (GlcCer) in lysosomes, particularly of tissue macrophages. As a result, GlcCer accumulates, which, to a certain extent, is converted to its deacylated form, glucosylsphingosine (GlcSph), by lysosomal acid ceramidase. The inability of mutant GCase to degrade GlcSph further promotes its accumulation. The amount of mutant GCase in lysosomes depends on the amount of mutant ER enzyme that shuttles to them. In the case of many mutant GCase forms, the enzyme is largely misfolded in the ER. Only a fraction correctly folds and is subsequently trafficked to the lysosomes, while the rest of the misfolded mutant GCase protein undergoes ER-associated degradation (ERAD). The retention of misfolded mutant GCase in the ER induces ER stress, which evokes a stress response known as the unfolded protein response (UPR). GD is remarkably heterogeneous in clinical manifestation, including the variant without CNS involvement (type 1), and acute and subacute neuronopathic variants (types 2 and 3). The present review discusses animal models developed to study the molecular and cellular mechanisms underlying GD.
Keywords: ER stress; GBA1; glucocerebrosidase (GCase); glucosylceramide (GlcCer); inflammation; knockin animals; knockout animals; misfolding; unfolded protein response (UPR).
Conflict of interest statement
The authors declare no conflict of interest.
Figures







Similar articles
-
Lysosomal functions and dysfunctions: Molecular and cellular mechanisms underlying Gaucher disease and its association with Parkinson disease.Adv Drug Deliv Rev. 2022 Aug;187:114402. doi: 10.1016/j.addr.2022.114402. Epub 2022 Jun 25. Adv Drug Deliv Rev. 2022. PMID: 35764179 Review.
-
Elevated glucosylsphingosine in Gaucher disease induced pluripotent stem cell neurons deregulates lysosomal compartment through mammalian target of rapamycin complex 1.Stem Cells Transl Med. 2021 Jul;10(7):1081-1094. doi: 10.1002/sctm.20-0386. Epub 2021 Mar 3. Stem Cells Transl Med. 2021. PMID: 33656802 Free PMC article.
-
Glucosylceramide and Glucosylsphingosine Quantitation by Liquid Chromatography-Tandem Mass Spectrometry to Enable In Vivo Preclinical Studies of Neuronopathic Gaucher Disease.Anal Chem. 2017 Aug 15;89(16):8288-8295. doi: 10.1021/acs.analchem.7b01442. Epub 2017 Jul 26. Anal Chem. 2017. PMID: 28686011
-
Unfolded protein response in Gaucher disease: from human to Drosophila.Orphanet J Rare Dis. 2013 Sep 11;8:140. doi: 10.1186/1750-1172-8-140. Orphanet J Rare Dis. 2013. PMID: 24020503 Free PMC article.
-
New Directions in Gaucher Disease.Hum Mutat. 2016 Nov;37(11):1121-1136. doi: 10.1002/humu.23056. Epub 2016 Aug 21. Hum Mutat. 2016. PMID: 27449603 Review.
Cited by
-
Advancements in Viral Gene Therapy for Gaucher Disease.Genes (Basel). 2024 Mar 15;15(3):364. doi: 10.3390/genes15030364. Genes (Basel). 2024. PMID: 38540423 Free PMC article. Review.
-
A Comparative Biochemical and Pathological Evaluation of Brain Samples from Knock-In Murine Models of Gaucher Disease.Int J Mol Sci. 2024 Feb 2;25(3):1827. doi: 10.3390/ijms25031827. Int J Mol Sci. 2024. PMID: 38339105 Free PMC article.
-
Zebrafish navigating the metabolic maze: insights into human disease - assets, challenges and future implications.J Diabetes Metab Disord. 2024 Dec 16;24(1):3. doi: 10.1007/s40200-024-01539-8. eCollection 2025 Jun. J Diabetes Metab Disord. 2024. PMID: 39697864 Review.
-
Diet and Nutrients in Rare Neurological Disorders: Biological, Biochemical, and Pathophysiological Evidence.Nutrients. 2024 Sep 15;16(18):3114. doi: 10.3390/nu16183114. Nutrients. 2024. PMID: 39339713 Free PMC article. Review.
References
-
- Lieberman R.L., Wustman B.A., Huertas P., Powe A.C., Jr., Pine C.W., Khanna R., Schlossmacher M.G., Ringe D., Petsko G.A. Structure of acid beta-glucosidase with pharmacological chaperone provides insight into Gaucher disease. Nat. Chem. Biol. 2007;3:101–107. doi: 10.1038/nchembio850. - DOI - PubMed
-
- Brumshtein B., Aguilar-Moncayo M., García-Moreno M.I., Ortiz Mellet C., García Fernández J.M., Silman I., Shaaltiel Y., Aviezer D., Sussman J.L., Futerman A.H. 6-Amino-6-deoxy-5,6-di-N-(N’-octyliminomethylidene)nojirimycin: Synthesis, biological evaluation, and crystal structure in complex with acid beta-glucosidase. Chembiochem. 2009;10:1480–1485. doi: 10.1002/cbic.200900142. - DOI - PubMed
-
- Kallemeijn W.W., Witte M.D., Voorn-Brouwer T.M., Walvoort M.T., Li K.Y., Codée J.D., van der Marel G.A., Boot R.G., Overkleeft H.S., Aerts J.M. A sensitive gel-based method combining distinct cyclophellitol-based probes for the identification of acid/base residues in human retaining β-glucosidases. J. Biol. Chem. 2014;289:35351–35362. doi: 10.1074/jbc.M114.593376. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
