

Aicardi-Goutières Syndrome with Congenital Glaucoma Caused by Novel TREX1 Mutation
- PMID: 38003924
- PMCID: PMC10672266
- DOI: 10.3390/jpm13111609
Aicardi-Goutières Syndrome with Congenital Glaucoma Caused by Novel TREX1 Mutation
Abstract
Background: Aicardi-Goutières syndrome (AGS) is a rare genetic disorder characterized by microcephaly, white matter lesions, numerous intracranial calcifications, chilblain skin lesions and high levels of interferon-α (IFN-α) in the cerebrospinal fluid (CSF). However, ocular involvement is reported significantly less frequently.
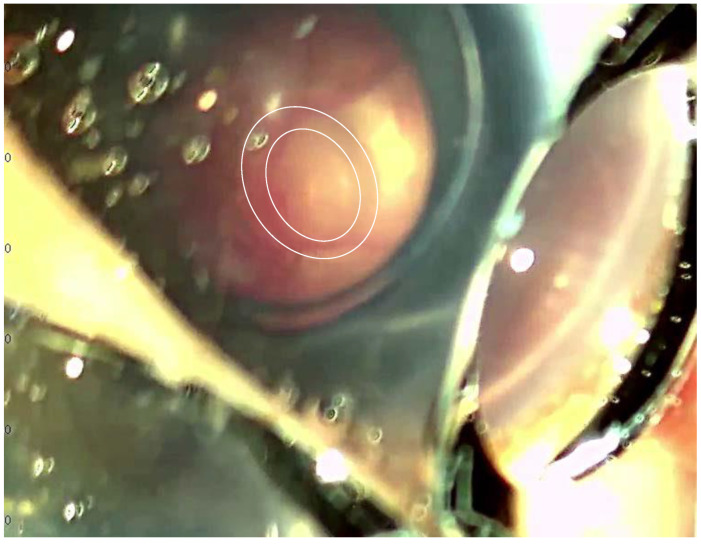
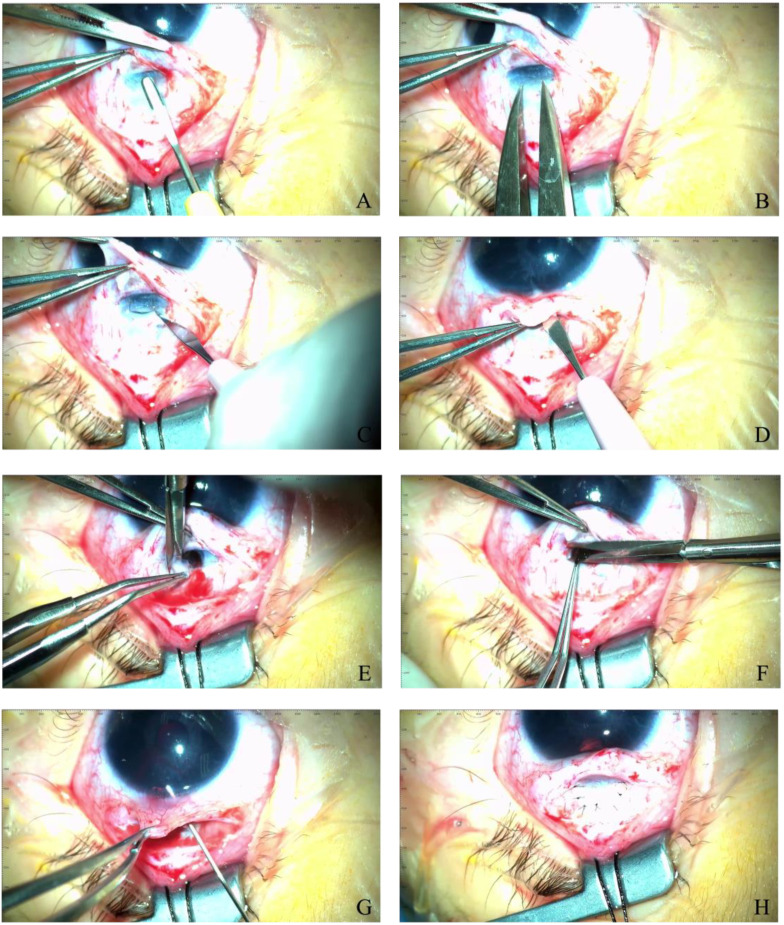
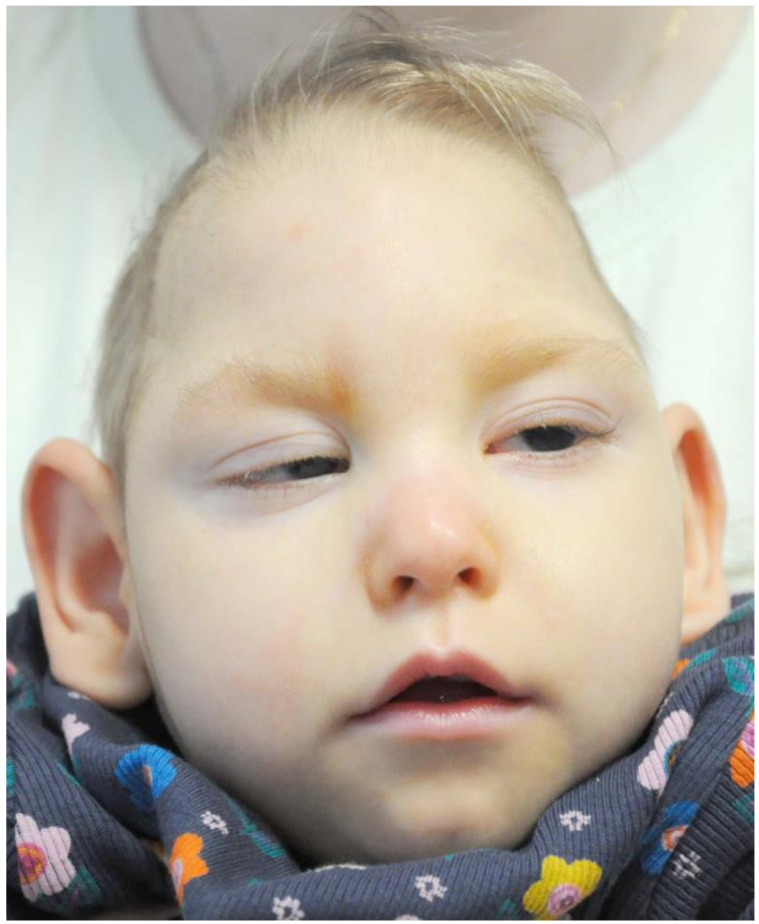
Case presentation: We present a case of a neonate with hypotrophy, microcephaly, frostbite-like skin lesions, thrombocytopenia, elevated liver enzymes and hepatosplenomegaly. Magnetic resonance imaging (MRI) of the brain showed multiple foci of calcification, white matter changes, cerebral atrophy, and atrophic dilatation of the ventricular system. The inflammatory parameters were not elevated, and the infectious etiology was excluded. Instead, elevated levels of IFN-α in the serum were detected. Based on the related clinical symptoms, imaging and test findings, the diagnosis of AGS was suspected. Genetic testing revealed two pathogenic mutations, c.490C>T and c.222del (novel mutation), in the three prime repair exonuclease 1 (TREX1) gene, confirming AGS type 1 (AGS1). An ophthalmologic examination of the child at 10 months of age revealed an impaired pupillary response to light, a corneal haze with Haab lines in the right eye (RE), pale optic nerve discs and neuropathy in both eyes (OU). The intraocular pressure (IOP) was 51 mmHg in the RE and 49 in the left eye (LE). The flash visual evoked potential (FVEP) showed prolonged P2 latencies of up to 125% in the LE and reduced amplitudes of up to approximately 10% OU. This girl was diagnosed with congenital glaucoma, and it was managed with a trabeculectomy with a basal iridectomy of OU, resulting in a reduction and stabilization in the IOP to 12 mmHg in the RE and 10 mmHg in the LE without any hypotensive eyedrops.
Conclusions: We present the clinical characteristics, electrophysiological and imaging findings, as well as the genetic test results of a patient with AGS1. Our case contributes to the extended ophthalmic involvement of the pathogenic c.490C>T and c.222del mutations in TREX1.
Keywords: Aicardi–Goutières syndrome; TREX1; congenital glaucoma; interferon alpha; type 1 interferonopathies.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Case Report: Aicardi-Goutières Syndrome Caused by Novel TREX1 Variants.Front Pediatr. 2021 Apr 28;9:634281. doi: 10.3389/fped.2021.634281. eCollection 2021. Front Pediatr. 2021. PMID: 33996686 Free PMC article.
-
[Clinical and genetic analysis of a family with Aicardi-Goutières syndrome and literature review].Zhonghua Er Ke Za Zhi. 2014 Nov;52(11):822-7. Zhonghua Er Ke Za Zhi. 2014. PMID: 25582466 Review. Chinese.
-
A case of Aicardi-Goutières syndrome caused by TREX1 gene mutation.BMC Pregnancy Childbirth. 2023 Feb 22;23(1):124. doi: 10.1186/s12884-023-05436-5. BMC Pregnancy Childbirth. 2023. PMID: 36814213 Free PMC article.
-
Analysis of clinical characteristics of children with Aicardi-Goutieres syndrome in China.World J Pediatr. 2022 Jul;18(7):490-497. doi: 10.1007/s12519-022-00545-1. Epub 2022 May 12. World J Pediatr. 2022. PMID: 35551623 Free PMC article.
-
Aicardi-Goutieres syndrome, a rare neurological disease in children: a new autoimmune disorder?Autoimmun Rev. 2013 Feb;12(4):506-9. doi: 10.1016/j.autrev.2012.08.012. Epub 2012 Aug 24. Autoimmun Rev. 2013. PMID: 22940555 Review.
Cited by
-
High Tumor Mutation Burden (TMB) and a Novel Somatic Mutation in the TREX1 Gene in a Patient with Aggressive and Refractory High-Grade B-Cell Lymphoma: A Case Report.Int J Mol Sci. 2025 Mar 24;26(7):2926. doi: 10.3390/ijms26072926. Int J Mol Sci. 2025. PMID: 40243506 Free PMC article.
References
-
- Crow Y.J., Chase D.S., Lowenstein Schmidt J., Szynkiewicz M., Forte G.M., Gornall H.L., Oojageer A., Anderson B., Pizzino A., Helman G., et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A. 2015;2:296–312. doi: 10.1002/ajmg.a.36887. - DOI - PMC - PubMed
-
- Abe J., Nakamura K., Nishikomori R., Kato M., Mitsuiki N., Izawa K., Awaya T., Kawai T., Yasumi T., Toyoshima I., et al. A nationwide survey of Aicardi-Goutières syndrome patients identifies a strong association between dominant TREX1 mutations and chilblain lesions: Japanese cohort study. Rheumatology. 2014;3:448–458. doi: 10.1093/rheumatology/ket372. - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
