

Phylogenetic Analysis and Emerging Drug Resistance against Different Nucleoside Analogues in Hepatitis B Virus Positive Patients
- PMID: 38004634
- PMCID: PMC10673510
- DOI: 10.3390/microorganisms11112622
Phylogenetic Analysis and Emerging Drug Resistance against Different Nucleoside Analogues in Hepatitis B Virus Positive Patients
Abstract
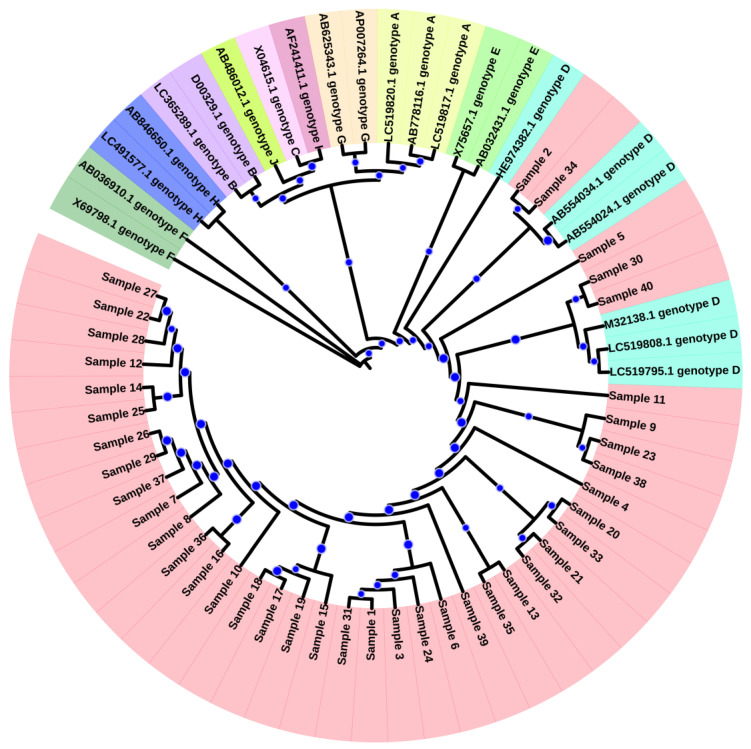
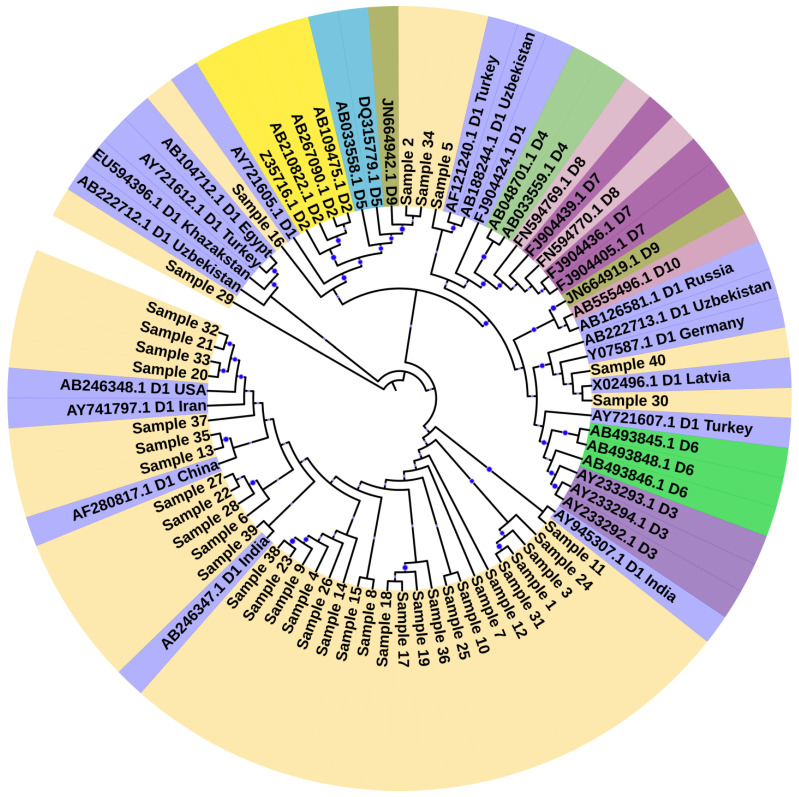
Several nucleotide analogues have been approved for use in treating hepatitis B virus (HBV) infection. Long-term exposure to therapy leads to the emergence of mutations within the HBV DNA polymerase gene, resulting in drug resistance, a major factor contributing to therapy failure. Chronic HBV patients from the Khyber Pakhtunkhwa province, Pakistan, who had completed 6 months of therapy participated in this study. Samples were collected from 60 patients. In this study, the entire reverse transcriptase domain of the HBV polymerase gene was amplified using nested polymerase chain reaction and sequenced. Drug-resistant mutations were detected in nine (22.5%) patients. All of these patients had lamivudine-resistant mutations (rtM204V + L180M), while seven individuals (17.5%) had both lamivudine- plus entecavir-resistant mutations (L180M + M204V + S202G). N236T, a mutation that gives rise to tenofovir and adefovir resistance, was observed in two (5%) patients. T184A, a partial drug-resistant mutation to entecavir, was found in five (12.5%) patients. Furthermore, other genotypic variants (100%) and vaccine escape mutations (5%) were additionally observed. Moreover, pN459Y (35%), pN131D (20%), pL231S (20%), pP130Q (17.5%), pS189Q (12.5%), pP161S (5%), pH160P (2.5%), pT322S (2.5%), and pA223S (2.5%) mutations in the polymerase gene, as well as sA166V (17.5%), sQ181K (12.5%), sV184R (7.5%), sA17E (5%), sP153S/K (5%), sW156C (5%), sC76Y (2.5%), and S132F (2.5%) mutations in the small surface gene, were identified for the first time in this study. Phylogenetic analysis showed that genotype D was predominant amongst the HBV carriers. Subtype D1 was found in most patients, while two patients were subtype D9. These novel findings may contribute to the body of knowledge and have clinical significance for treating and curing HBV infections in Pakistan.
Keywords: genotypes; hepatitis B virus; mutations; phylogenetic analysis; polymerase gene; surface gene.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Investigation into drug-resistant mutations of HBV from 845 nucleoside/nucleotide analogue-naive Chinese patients with chronic HBV infection.Antivir Ther. 2015;20(2):141-7. doi: 10.3851/IMP2813. Epub 2014 Jul 3. Antivir Ther. 2015. PMID: 24992206
-
Hepatitis B virus mutation pattern rtL180M+A181C+M204V may contribute to entecavir resistance in clinical practice.Emerg Microbes Infect. 2019;8(1):354-365. doi: 10.1080/22221751.2019.1584018. Emerg Microbes Infect. 2019. PMID: 30866789 Free PMC article.
-
Unsuccessful therapy with adefovir and entecavir-tenofovir in a patient with chronic hepatitis B infection with previous resistance to lamivudine: a fourteen-year evolution of hepatitis B virus mutations.BMC Infect Dis. 2011 Jun 22;11:178. doi: 10.1186/1471-2334-11-178. BMC Infect Dis. 2011. PMID: 21696601 Free PMC article.
-
Nucleoside analogues for chronic hepatitis B: antiviral efficacy and viral resistance.Am J Gastroenterol. 2002 Jul;97(7):1618-28. doi: 10.1111/j.1572-0241.2002.05819.x. Am J Gastroenterol. 2002. PMID: 12135009 Review.
-
Treatment of chronic hepatitis B: Evolution over two decades.J Gastroenterol Hepatol. 2011 Jan;26 Suppl 1:138-43. doi: 10.1111/j.1440-1746.2010.06545.x. J Gastroenterol Hepatol. 2011. PMID: 21199525 Review.
Cited by
-
Hepatitis B Virus Prevalence among HIV-Uninfected People Living in Rural and Peri-Urban Areas in Botswana.Microorganisms. 2024 Jun 15;12(6):1207. doi: 10.3390/microorganisms12061207. Microorganisms. 2024. PMID: 38930589 Free PMC article.
References
-
- Dos Santos M.I.M.A., Pacheco S.R., Stocker A., Schinoni M.I., Paraná R., Reis M.G., Silva L.K. Mutations associated with drug resistance and prevalence of vaccine escape mutations in patients with chronic hepatitis B infection. J. Med. Virol. 2017;89:1811–1816. doi: 10.1002/jmv.24853. - DOI - PubMed
-
- Al-Sadeq D.W., Taleb S.A., Zaied R.E., Fahad S.M., Smatti M.K., Rizeq B.R., Al Thani A.A., Yassine H.M., Nasrallah G.K. Hepatitis B virus molecular epidemiology, host-virus interaction, coinfection, and laboratory diagnosis in the MENA region: An update. Pathogens. 2019;8:63. doi: 10.3390/pathogens8020063. - DOI - PMC - PubMed
-
- Hu J. Hepatitis B Virus in Human Diseases. Springer; Berlin/Heidelberg, Germany: 2016. Hepatitis B virus virology and replication; pp. 1–34.
Grants and funding
LinkOut - more resources
Full Text Sources
