

In Silico Evaluation, Phylogenetic Analysis, and Structural Modeling of the Class II Hydrophobin Family from Different Fungal Phytopathogens
- PMID: 38004644
- PMCID: PMC10672791
- DOI: 10.3390/microorganisms11112632
In Silico Evaluation, Phylogenetic Analysis, and Structural Modeling of the Class II Hydrophobin Family from Different Fungal Phytopathogens
Abstract
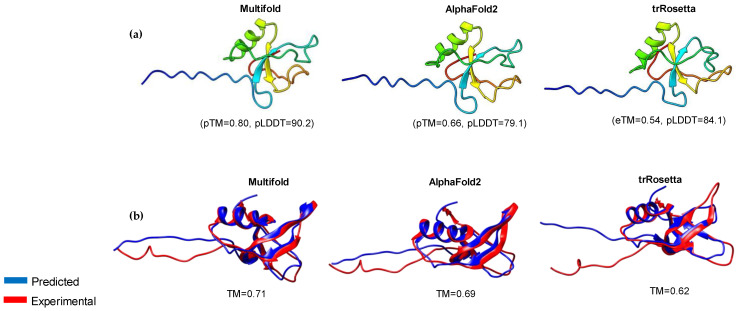
The class II hydrophobin group (HFBII) is an extracellular group of proteins that contain the HFBII domain and eight conserved cysteine residues. These proteins are exclusively secreted by fungi and have multiple functions with a probable role as effectors. In the present study, a total of 45 amino acid sequences of hydrophobin class II proteins from different phytopathogenic fungi were retrieved from the NCBI database. We used the integration of well-designed bioinformatic tools to characterize and predict their physicochemical parameters, novel motifs, 3D structures, multiple sequence alignment (MSA), evolution, and functions as effector proteins through molecular docking. The results revealed new features for these protein members. The ProtParam tool detected the hydrophobicity properties of all proteins except for one hydrophilic protein (KAI3335996.1). Out of 45 proteins, six of them were detected as GPI-anchored proteins by the PredGPI server. Different 3D structure templates with high pTM scores were designed by Multifold v1, AlphaFold2, and trRosetta. Most of the studied proteins were anticipated as apoplastic effectors and matched with the ghyd5 gene of Fusarium graminearum as virulence factors. A protein-protein interaction (PPI) analysis unraveled the molecular function of this group as GTP-binding proteins, while a molecular docking analysis detected a chitin-binding effector role. From the MSA analysis, it was observed that the HFBII sequences shared conserved 2 Pro (P) and 2 Gly (G) amino acids besides the known eight conserved cysteine residues. The evolutionary analysis and phylogenetic tree provided evidence of episodic diversifying selection at the branch level using the aBSREL tool. A detailed in silico analysis of this family and the present findings will provide a better understanding of the HFBII characters and evolutionary relationships, which could be very useful in future studies.
Keywords: computational annotation; effectors; evolution; homology modeling; hydrophobins.
Conflict of interest statement
The authors declare no conflict of interest.
Figures












References
-
- Quarantin A., Hadeler B., Kröger C., Schäfer W., Favaron F., Sella L., Martínez-Rocha A.L. Different hydrophobins of Fusarium graminearum are involved in hyphal growth, attachment, water-Air interface penetration and plant infection. Front. Microbiol. 2019;10:751. doi: 10.3389/fmicb.2019.00751. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
