

Genomic and Phenotypic Characterization of Shiga Toxin-Producing Escherichia albertii Strains Isolated from Wild Birds in a Major Agricultural Region in California
- PMID: 38004814
- PMCID: PMC10673567
- DOI: 10.3390/microorganisms11112803
Genomic and Phenotypic Characterization of Shiga Toxin-Producing Escherichia albertii Strains Isolated from Wild Birds in a Major Agricultural Region in California
Abstract
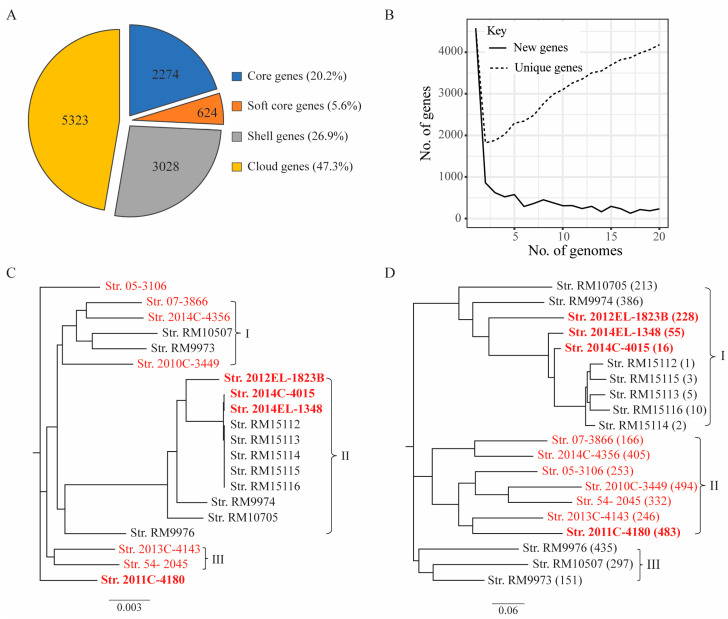
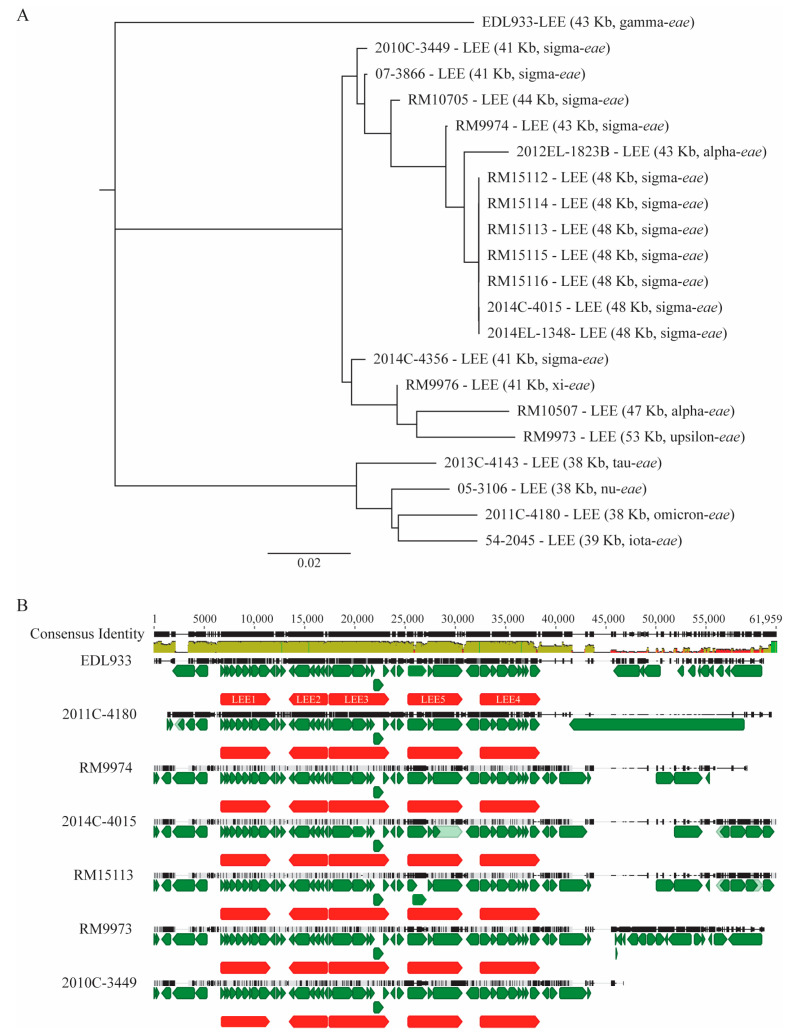
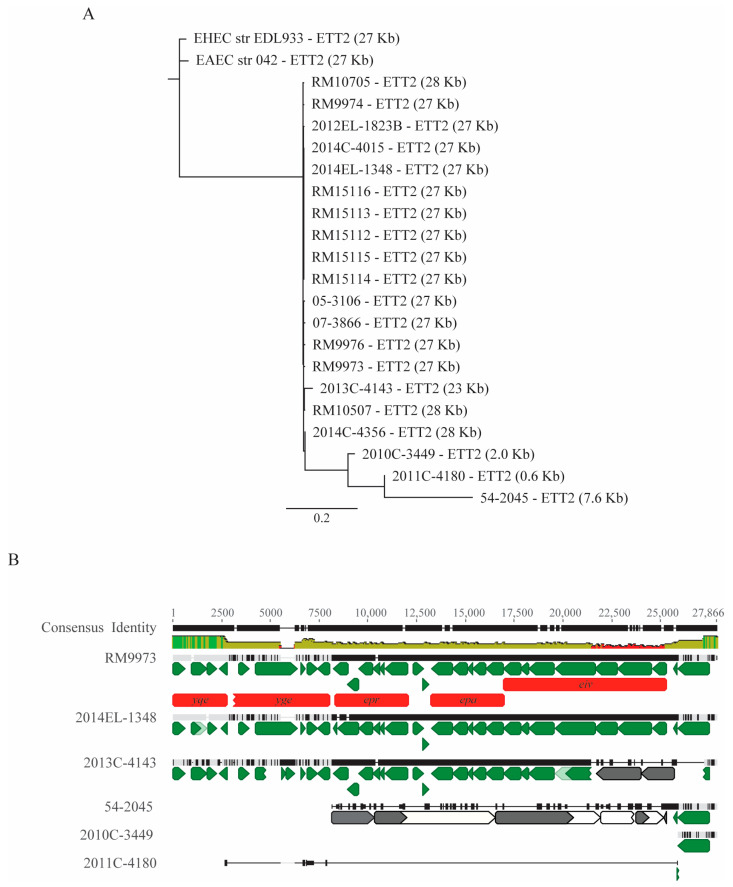
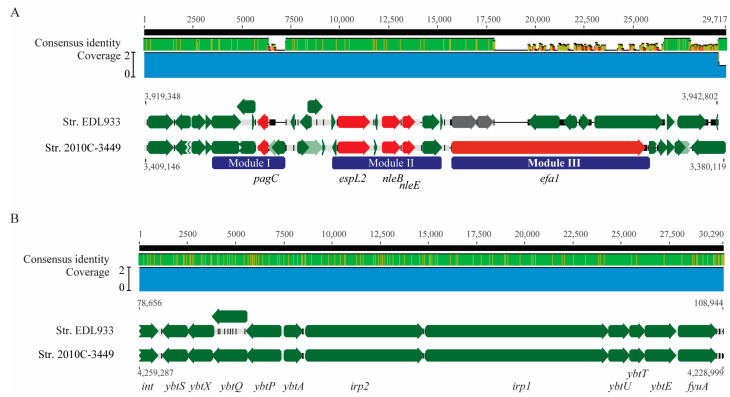
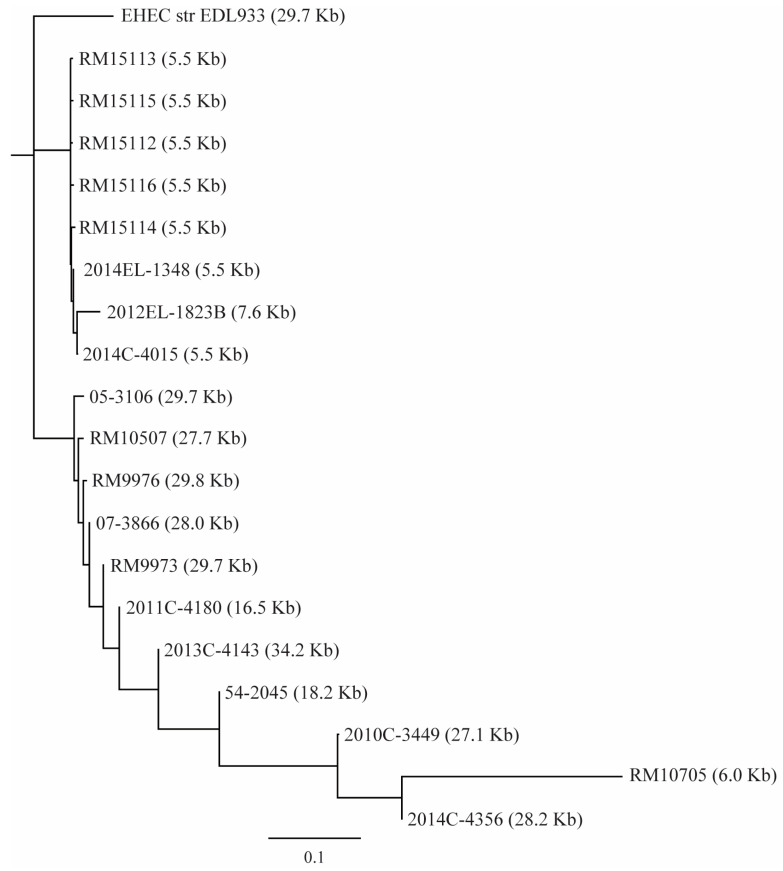
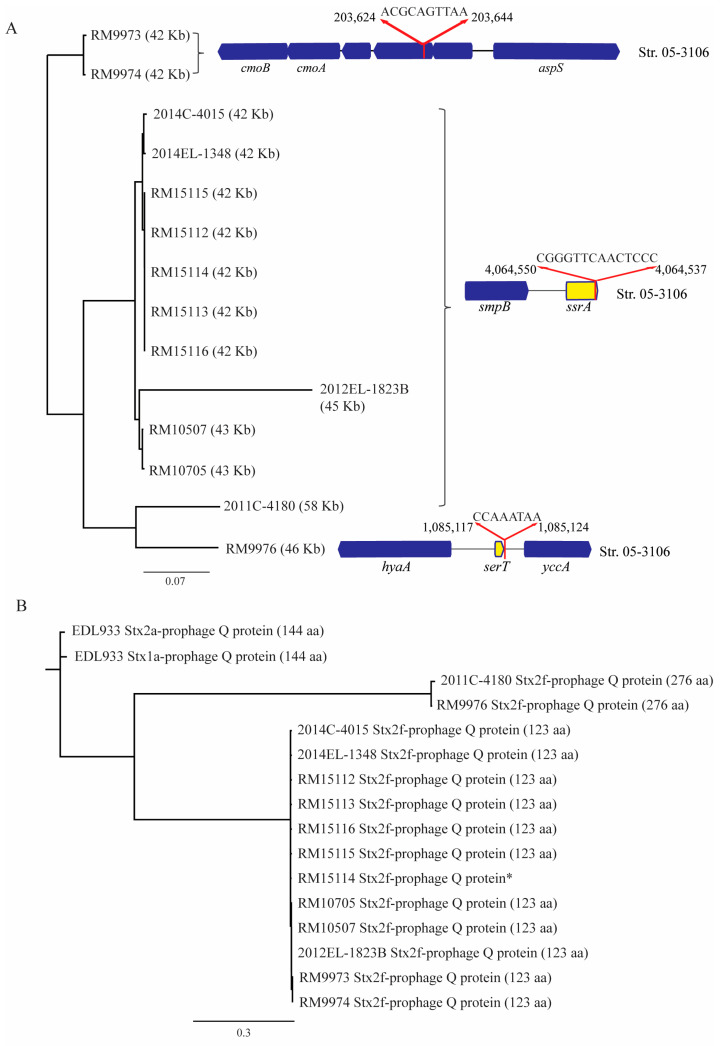
Escherichia albertii is an emerging foodborne pathogen. To better understand the pathogenesis and health risk of this pathogen, comparative genomics and phenotypic characterization were applied to assess the pathogenicity potential of E. albertii strains isolated from wild birds in a major agricultural region in California. Shiga toxin genes stx2f were present in all avian strains. Pangenome analyses of 20 complete genomes revealed a total of 11,249 genes, of which nearly 80% were accessory genes. Both core gene-based phylogenetic and accessory gene-based relatedness analyses consistently grouped the three stx2f-positive clinical strains with the five avian strains carrying ST7971. Among the three Stx2f-converting prophage integration sites identified, ssrA was the most common one. Besides the locus of enterocyte effacement and type three secretion system, the high pathogenicity island, OI-122, and type six secretion systems were identified. Substantial strain variation in virulence gene repertoire, Shiga toxin production, and cytotoxicity were revealed. Six avian strains exhibited significantly higher cytotoxicity than that of stx2f-positive E. coli, and three of them exhibited a comparable level of cytotoxicity with that of enterohemorrhagic E. coli outbreak strains, suggesting that wild birds could serve as a reservoir of E. albertii strains with great potential to cause severe diseases in humans.
Keywords: Escherichia albertii; Shiga toxin; cytolethal distending toxin; cytotoxicity; foodborne pathogen; intimin; pangenome; pathogenicity; phage integration; stx2f.
Conflict of interest statement
The findings and conclusions in this report are those of the author(s) and do not reflect the view of the Centers for Disease Control and Prevention, the Department of Health and Human Services, or the United States government. Furthermore, the use of any product names, trade names, images, or commercial sources is for identification purposes only, and does not imply endorsement or government sanction by the U.S. Department of Health and Human Services.
Figures
Similar articles
-
Conditional expression of flagellar motility, curli fimbriae, and biofilms in Shiga toxin- producing Escherichia albertii.Front Microbiol. 2024 Sep 9;15:1456637. doi: 10.3389/fmicb.2024.1456637. eCollection 2024. Front Microbiol. 2024. PMID: 39318426 Free PMC article.
-
Pathogenicity assessment of Shiga toxin-producing Escherichia coli strains isolated from wild birds in a major agricultural region in California.Front Microbiol. 2023 Sep 26;14:1214081. doi: 10.3389/fmicb.2023.1214081. eCollection 2023. Front Microbiol. 2023. PMID: 37822735 Free PMC article.
-
Comparative genomic and phenotypic analyses of the virulence potential in Shiga toxin-producing Escherichia coli O121:H7 and O121:H10.Front Cell Infect Microbiol. 2022 Nov 24;12:1043726. doi: 10.3389/fcimb.2022.1043726. eCollection 2022. Front Cell Infect Microbiol. 2022. PMID: 36506028 Free PMC article.
-
Escherichia albertii Pathogenesis.EcoSal Plus. 2020 Jun;9(1):10.1128/ecosalplus.ESP-0015-2019. doi: 10.1128/ecosalplus.ESP-0015-2019. EcoSal Plus. 2020. PMID: 32588811 Free PMC article. Review.
-
Toxins of Locus of Enterocyte Effacement-Negative Shiga Toxin-Producing Escherichia coli.Toxins (Basel). 2018 Jun 14;10(6):241. doi: 10.3390/toxins10060241. Toxins (Basel). 2018. PMID: 29903982 Free PMC article. Review.
Cited by
-
Conditional expression of flagellar motility, curli fimbriae, and biofilms in Shiga toxin- producing Escherichia albertii.Front Microbiol. 2024 Sep 9;15:1456637. doi: 10.3389/fmicb.2024.1456637. eCollection 2024. Front Microbiol. 2024. PMID: 39318426 Free PMC article.
-
Intestinal Carriage of Two Distinct stx2f-Carrying Escherichia coli Strains by a Child with Uncomplicated Diarrhea.Pathogens. 2024 Nov 15;13(11):1002. doi: 10.3390/pathogens13111002. Pathogens. 2024. PMID: 39599555 Free PMC article.
-
Genomic characterization of multidrug-resistant Escherichia albertii of fish origin-first isolation and insights into a potential food safety threat.Front Microbiol. 2025 Feb 27;16:1521202. doi: 10.3389/fmicb.2025.1521202. eCollection 2025. Front Microbiol. 2025. PMID: 40083789 Free PMC article.
-
Excess A-subunits of Shiga toxin 2a are produced in enterohemorrhagic Escherichia coli.Sci Rep. 2025 May 14;15(1):16712. doi: 10.1038/s41598-025-01342-2. Sci Rep. 2025. PMID: 40368985 Free PMC article.
-
Comparative analyses of persistence traits in Escherichia coli O157:H7 strains belonging to different clades including REPEXH01 and REPEXH02 strains.Front Microbiol. 2024 Dec 18;15:1501956. doi: 10.3389/fmicb.2024.1501956. eCollection 2024. Front Microbiol. 2024. PMID: 39744393 Free PMC article.
References
-
- Hyma K.E., Lacher D.W., Nelson A.M., Bumbaugh A.C., Janda J.M., Strockbine N.A., Young V.B., Whittam T.S. Evolutionary genetics of a new pathogenic Escherichia species: Escherichia albertii and related Shigella boydii strains. J. Bacteriol. 2005;187:619–628. doi: 10.1128/JB.187.2.619-628.2005. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
