

Caspase-8 activation by cigarette smoke induces pro-inflammatory cell death of human macrophages exposed to lipopolysaccharide
- PMID: 38007509
- PMCID: PMC10676397
- DOI: 10.1038/s41419-023-06318-6
Caspase-8 activation by cigarette smoke induces pro-inflammatory cell death of human macrophages exposed to lipopolysaccharide
Abstract
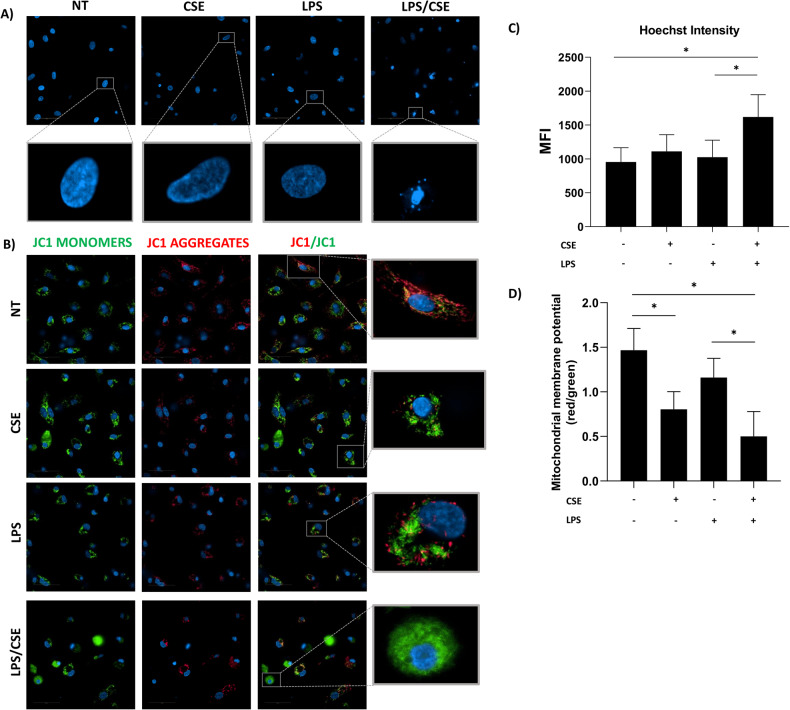
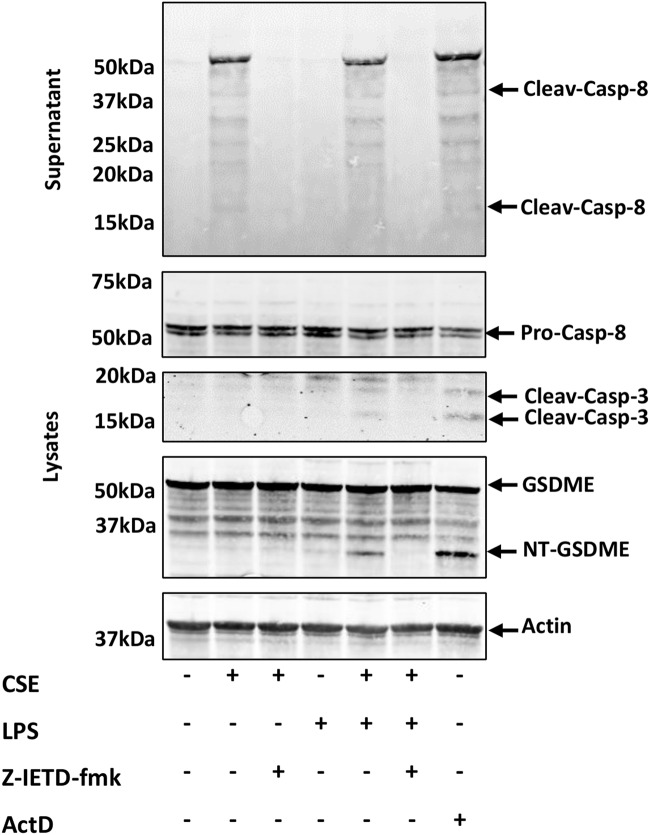
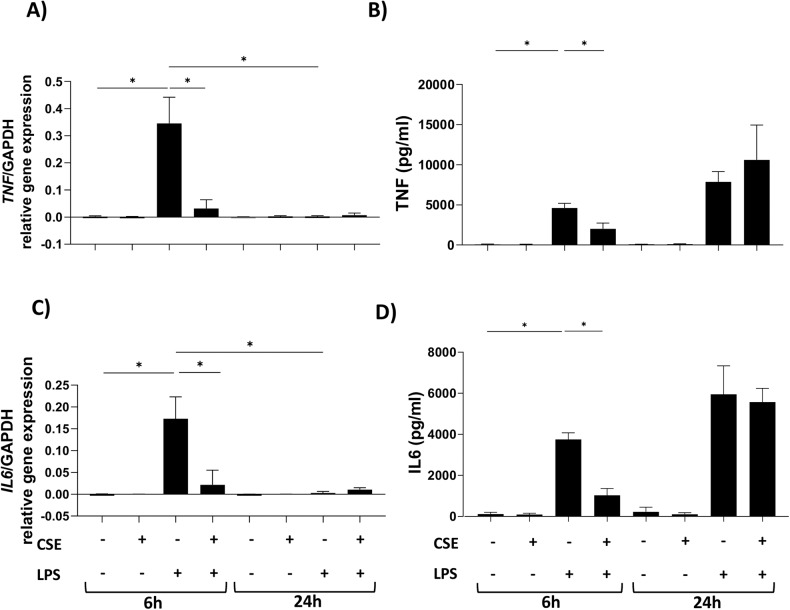
Cigarette smoking impairs the lung innate immune response making smokers more susceptible to infections and severe symptoms. Dysregulation of cell death is emerging as a key player in chronic inflammatory conditions. We have recently reported that short exposure of human monocyte-derived macrophages (hMDMs) to cigarette smoke extract (CSE) altered the TLR4-dependent response to lipopolysaccharide (LPS). CSE caused inhibition of the MyD88-dependent inflammatory response and activation of TRIF/caspase-8/caspase-1 pathway leading to Gasdermin D (GSDMD) cleavage and increased cell permeability. Herein, we tested the hypothesis that activation of caspase-8 by CSE increased pro-inflammatory cell death of LPS-stimulated macrophages. To this purpose, we measured apoptotic and pyroptotic markers as well as the expression/release of pro-inflammatory mediators in hMDMs exposed to LPS and CSE, alone or in combination, for 6 and 24 h. We show that LPS/CSE-treated hMDMs, but not cells treated with CSE or LPS alone, underwent lytic cell death (LDH release) and displayed apoptotic features (activation of caspase-8 and -3/7, nuclear condensation, and mitochondrial membrane depolarization). Moreover, the negative regulator of caspase-8, coded by CFLAR gene, was downregulated by CSE. Activation of caspase-3 led to Gasdermin E (GSDME) cleavage. Notably, lytic cell death caused the release of the damage-associated molecular patterns (DAMPs) heat shock protein-60 (HSP60) and S100A8/A9. This was accompanied by an impaired inflammatory response resulting in inhibited and delayed release of IL6 and TNF. Of note, increased cleaved caspase-3, higher levels of GSDME and altered expression of cell death-associated genes were found in alveolar macrophages of smoker subjects compared to non-smoking controls. Overall, our findings show that CSE sensitizes human macrophages to cell death by promoting pyroptotic and apoptotic pathways upon encountering LPS. We propose that while the delayed inflammatory response may result in ineffective defenses against infections, the observed cell death associated with DAMP release may contribute to establish chronic inflammation. CS exposure sensitizes human macrophages to pro-inflammatory cell death. Upon exposure to LPS, CS inhibits the TLR4/MyD88 inflammatory response, downregulating the pro-inflammatory genes TNF and IL6 and the anti-apoptotic gene CFLAR, known to counteract caspase-8 activity. CS enhances caspase-8 activation through TLR4/TRIF, with a partial involvement of RIPK1, resulting on the activation of caspase-1/GSDMD axis leading to increased cell permeability and DAMP release through gasdermin pores [19]. At later timepoints caspase-3 becomes strongly activated by caspase-8 triggering apoptotic events which are associated with mitochondrial membrane depolarization, gasdermin E cleavage and secondary necrosis with consequent massive DAMP release.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
