

Robust mapping of spatiotemporal trajectories and cell-cell interactions in healthy and diseased tissues
- PMID: 38007580
- PMCID: PMC10676408
- DOI: 10.1038/s41467-023-43120-6
Robust mapping of spatiotemporal trajectories and cell-cell interactions in healthy and diseased tissues
Abstract
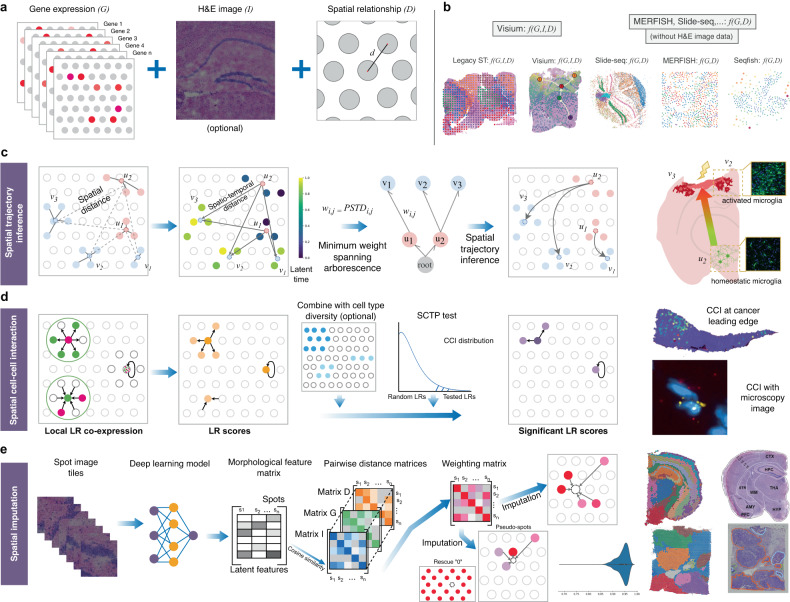
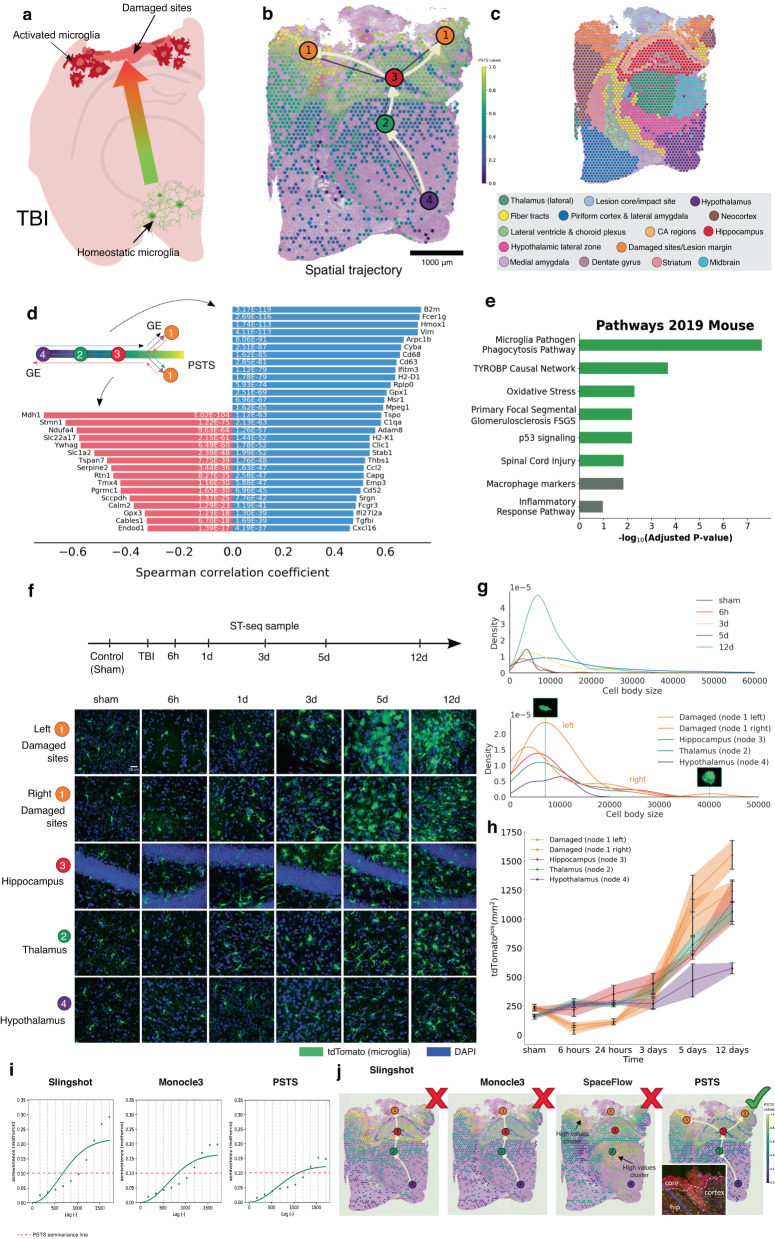
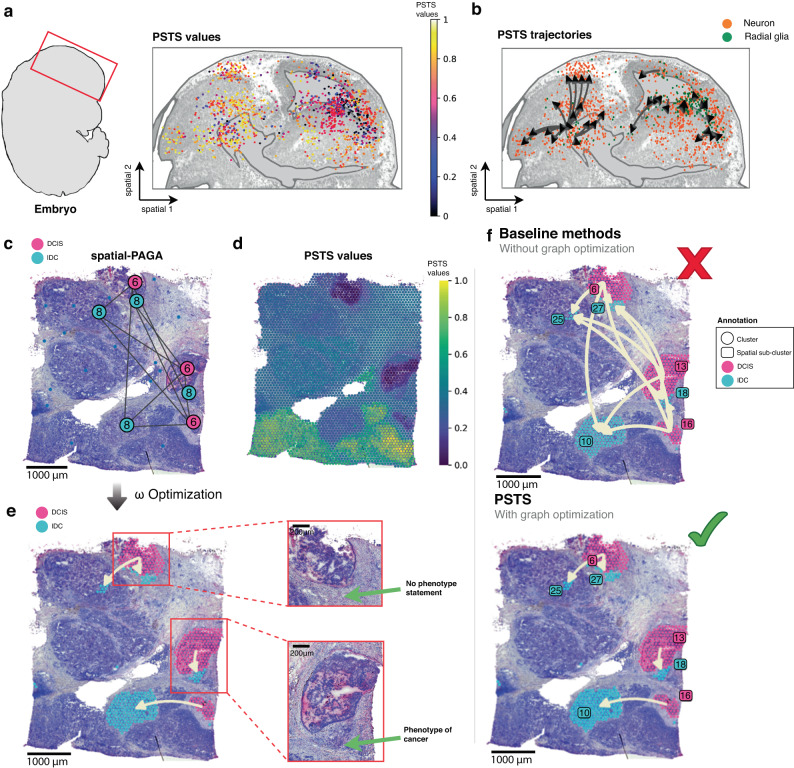
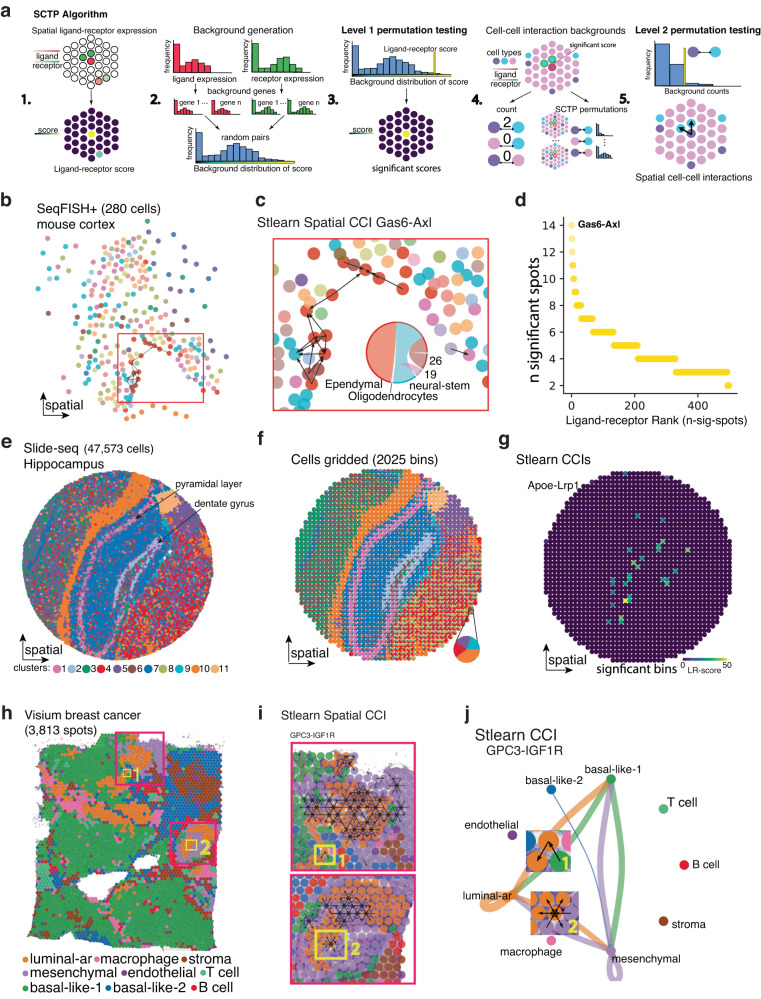
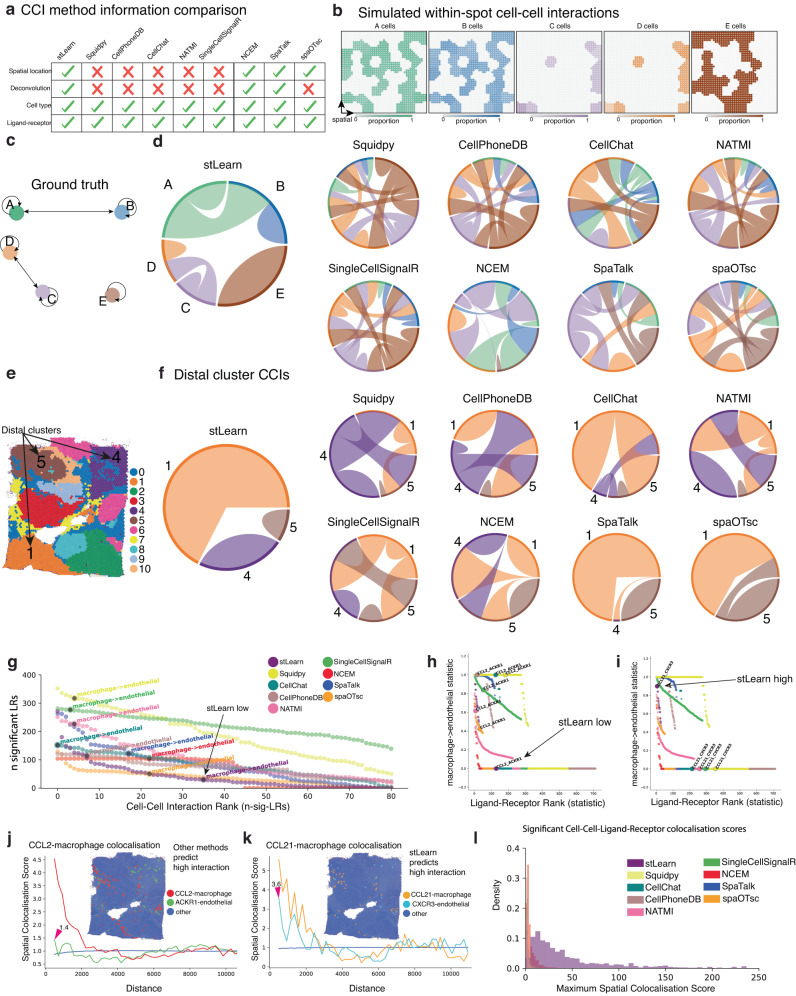
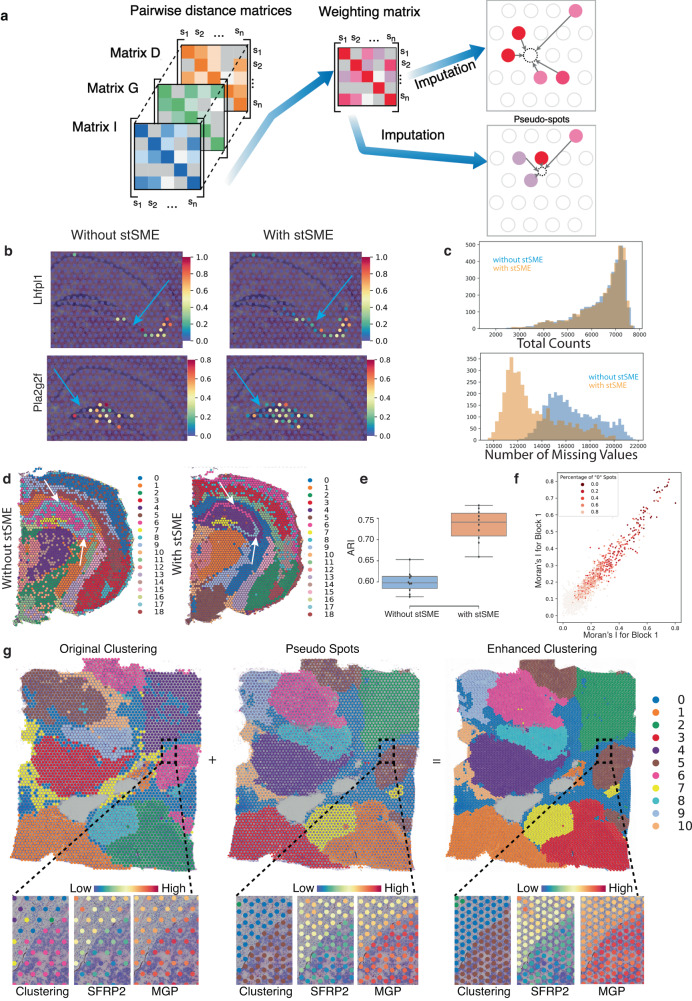
Spatial transcriptomics (ST) technologies generate multiple data types from biological samples, namely gene expression, physical distance between data points, and/or tissue morphology. Here we developed three computational-statistical algorithms that integrate all three data types to advance understanding of cellular processes. First, we present a spatial graph-based method, pseudo-time-space (PSTS), to model and uncover relationships between transcriptional states of cells across tissues undergoing dynamic change (e.g. neurodevelopment, brain injury and/or microglia activation, and cancer progression). We further developed a spatially-constrained two-level permutation (SCTP) test to study cell-cell interaction, finding highly interactive tissue regions across thousands of ligand-receptor pairs with markedly reduced false discovery rates. Finally, we present a spatial graph-based imputation method with neural network (stSME), to correct for technical noise/dropout and increase ST data coverage. Together, the algorithms that we developed, implemented in the comprehensive and fast stLearn software, allow for robust interrogation of biological processes within healthy and diseased tissues.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
- 2001514/Department of Health | National Health and Medical Research Council (NHMRC)
- GNT2008928/Department of Health | National Health and Medical Research Council (NHMRC)
- 1124503/Department of Health | National Health and Medical Research Council (NHMRC)
- 1163835/Department of Health | National Health and Medical Research Council (NHMRC)
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
