

Clinical and imaging predictors of late-onset GM2 gangliosidosis: A scoping review
- PMID: 38009419
- PMCID: PMC10791033
- DOI: 10.1002/acn3.51947
Clinical and imaging predictors of late-onset GM2 gangliosidosis: A scoping review
Abstract
Objective: Late-onset GM2 gangliosidosis (LOGG) subtypes late-onset Tay-Sachs (LOTS) and Sandhoff disease (LOSD) are ultra-rare neurodegenerative lysosomal storage disorders presenting with weakness, ataxia, and neuropsychiatric symptoms. Previous studies considered LOTS and LOSD clinically indistinguishable; recent studies have challenged this. We performed a scoping review to ascertain whether imaging and clinical features may differentiate these diseases.
Methods: We examined MEDLINE/non-MEDLINE databases up to May 2022. Articles reporting brain imaging findings in genetically/enzymatically confirmed LOGG, symptom onset at age ≥ 10 years (or evaluated at least once ≥18 years) were included, yielding 170 LOGG patients (LOTS = 127, LOSD = 43) across 68 papers. We compared LOTS versus LOSD and performed regression analyses. Results were corrected for multiple comparisons.
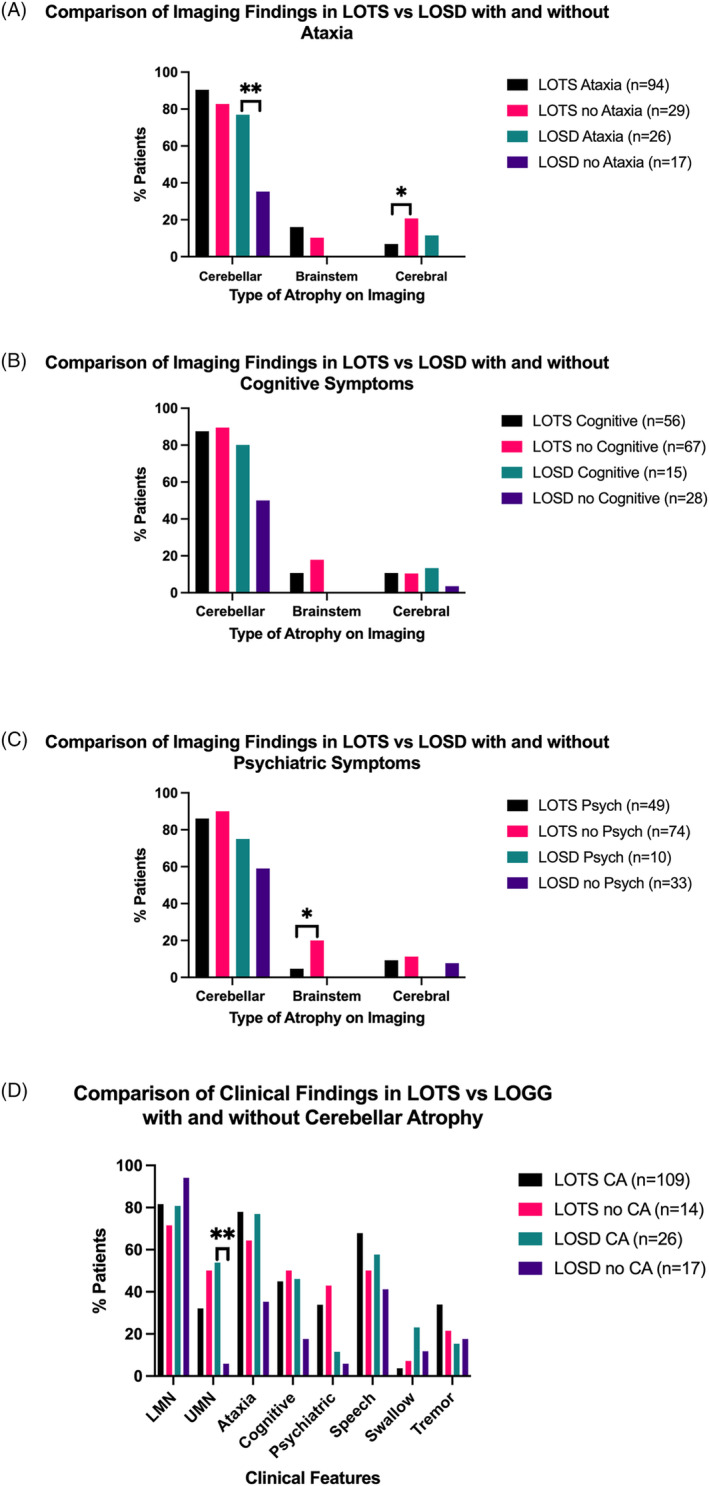
Results: Age of onset was lower in LOTS versus LOSD (17.9 ± 8.2 vs. 23.9 ± 14.4 years, p = 0.017), although disease duration was similar (p = 0.34). LOTS more commonly had psychosis/bipolar symptoms (35.0% vs. 9.30%, p = 0.011) but less frequent swallowing problems (4.10% vs. 18.60%, p = 0.041). Cerebellar atrophy was more common in LOTS (89.0%) versus LOSD (60.5%), p < 0.0001, with more severe atrophy in LOTS (p = 0.0005). Brainstem atrophy was documented only in LOTS (14.2%). Independent predictors of LOTS versus LOSD (odds ratio [95% confidence interval]) included the presence of psychosis/bipolar symptoms (4.95 [1.59-19.52], p = 0.011), no swallowing symptoms (0.16 [0.036-0.64], p = 0.011), and cerebellar atrophy (5.81 [2.10-17.08], p = 0.0009). Lower age of onset (0.96 [0.93-1.00], p = 0.075) and tremor (2.50 [0.94-7.43], p = 0.078) were marginally statistically significant but felt relevant to include in the model.
Interpretation: These data suggest significant differences in symptomatology, disease course, and imaging findings between LOTS and LOSD.
© 2023 The Authors. Annals of Clinical and Translational Neurology published by Wiley Periodicals LLC on behalf of American Neurological Association.
Figures



Similar articles
-
Late-onset GM2 gangliosidosis: magnetic resonance imaging, diffusion tensor imaging, and correlational fiber tractography differentiate Tay-Sachs and Sandhoff diseases.J Neurol. 2025 Apr 23;272(5):355. doi: 10.1007/s00415-025-13091-3. J Neurol. 2025. PMID: 40266357 Free PMC article.
-
Brainstem Substructure Atrophy in Late-Onset GM2-Gangliosidosis Imaging Using Automated Segmentation.Cerebellum. 2025 Feb 18;24(2):50. doi: 10.1007/s12311-025-01803-4. Cerebellum. 2025. PMID: 39966257 Free PMC article.
-
Magnetic resonance imaging and spectroscopy in late-onset GM2-gangliosidosis.Mol Genet Metab. 2021 Aug;133(4):386-396. doi: 10.1016/j.ymgme.2021.06.008. Epub 2021 Jun 24. Mol Genet Metab. 2021. PMID: 34226107 Free PMC article.
-
The natural history of juvenile or subacute GM2 gangliosidosis: 21 new cases and literature review of 134 previously reported.Pediatrics. 2006 Nov;118(5):e1550-62. doi: 10.1542/peds.2006-0588. Epub 2006 Oct 2. Pediatrics. 2006. PMID: 17015493 Free PMC article. Review.
-
[Molecular pathogenesis and therapeutic approach of GM2 gangliosidosis].Yakugaku Zasshi. 2013;133(2):269-74. doi: 10.1248/yakushi.12-00199. Yakugaku Zasshi. 2013. PMID: 23370522 Review. Japanese.
Cited by
-
Tay-Sachs and Sandhoff Diseases: Diffusion tensor imaging and correlational fiber tractography findings differentiate late-onset GM2 Gangliosidosis.medRxiv [Preprint]. 2024 Dec 16:2024.12.13.24318793. doi: 10.1101/2024.12.13.24318793. medRxiv. 2024. Update in: J Neurol. 2025 Apr 23;272(5):355. doi: 10.1007/s00415-025-13091-3. PMID: 39802759 Free PMC article. Updated. Preprint.
-
Late-onset GM2 gangliosidosis: magnetic resonance imaging, diffusion tensor imaging, and correlational fiber tractography differentiate Tay-Sachs and Sandhoff diseases.J Neurol. 2025 Apr 23;272(5):355. doi: 10.1007/s00415-025-13091-3. J Neurol. 2025. PMID: 40266357 Free PMC article.
-
Brainstem Substructure Atrophy in Late-Onset GM2-Gangliosidosis Imaging Using Automated Segmentation.Cerebellum. 2025 Feb 18;24(2):50. doi: 10.1007/s12311-025-01803-4. Cerebellum. 2025. PMID: 39966257 Free PMC article.
-
Clinical outcome assessments of disease burden and progression in late-onset GM2 gangliosidoses.Mol Genet Metab. 2024 Jul;142(3):108512. doi: 10.1016/j.ymgme.2024.108512. Epub 2024 Jun 6. Mol Genet Metab. 2024. PMID: 38870773 Free PMC article.
-
Deep Learning Cerebellar Magnetic Resonance Imaging Segmentation in Late-Onset GM2 Gangliosidosis: Implications for Phenotype.medRxiv [Preprint]. 2025 Apr 11:2025.04.08.25325262. doi: 10.1101/2025.04.08.25325262. medRxiv. 2025. PMID: 40297453 Free PMC article. Preprint.
References
-
- Toro C, Shirvan L, Tifft C. HEXA disorders. In: Adam MP, Everman DB, Mirzaa GM, et al., eds. GeneReviews(®). University of Washington; 1993. Copyright © 1993–2023, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All Rights Reserved.
-
- Jellinger K, Anzil AP, Seemann D, Bernheimer H. Adult GM2 gangliosidosis masquerading as slowly progressive muscular atrophy: motor neuron disease phenotype. Clin Neuropathol. 1982;1(1):31‐44. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
