

LipIDens: simulation assisted interpretation of lipid densities in cryo-EM structures of membrane proteins
- PMID: 38012131
- PMCID: PMC10682427
- DOI: 10.1038/s41467-023-43392-y
LipIDens: simulation assisted interpretation of lipid densities in cryo-EM structures of membrane proteins
Abstract
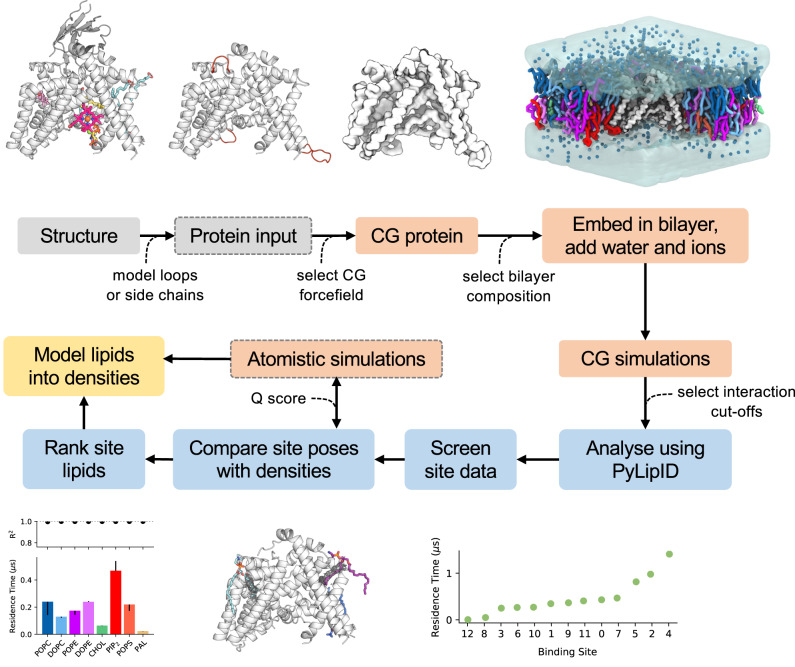
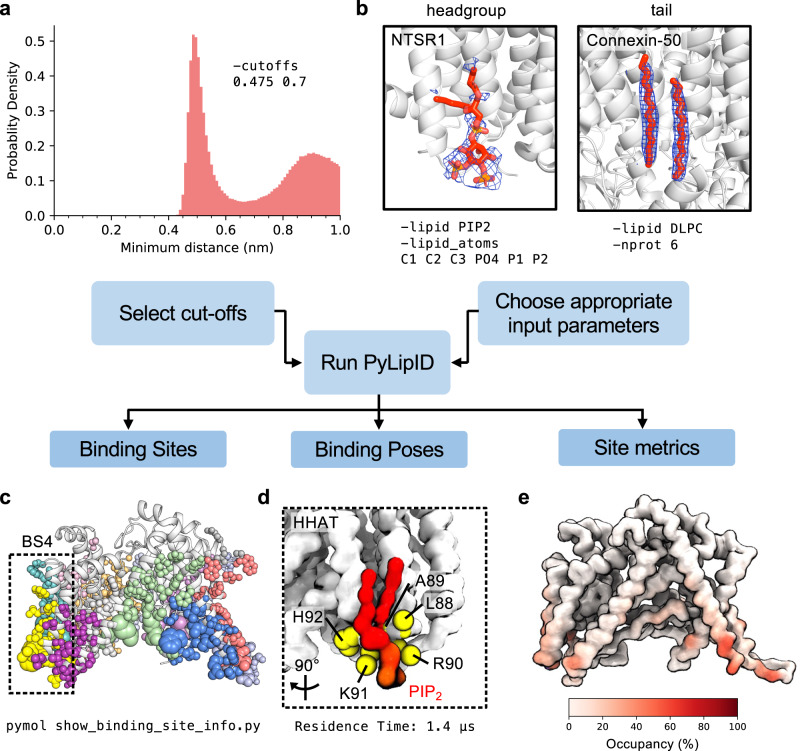
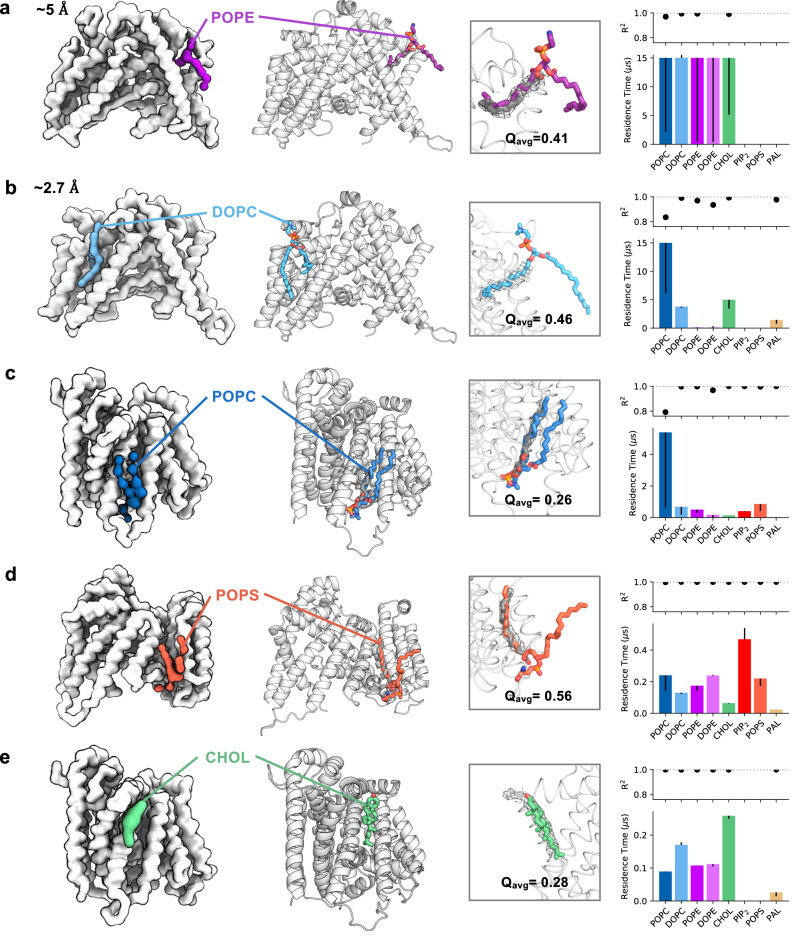
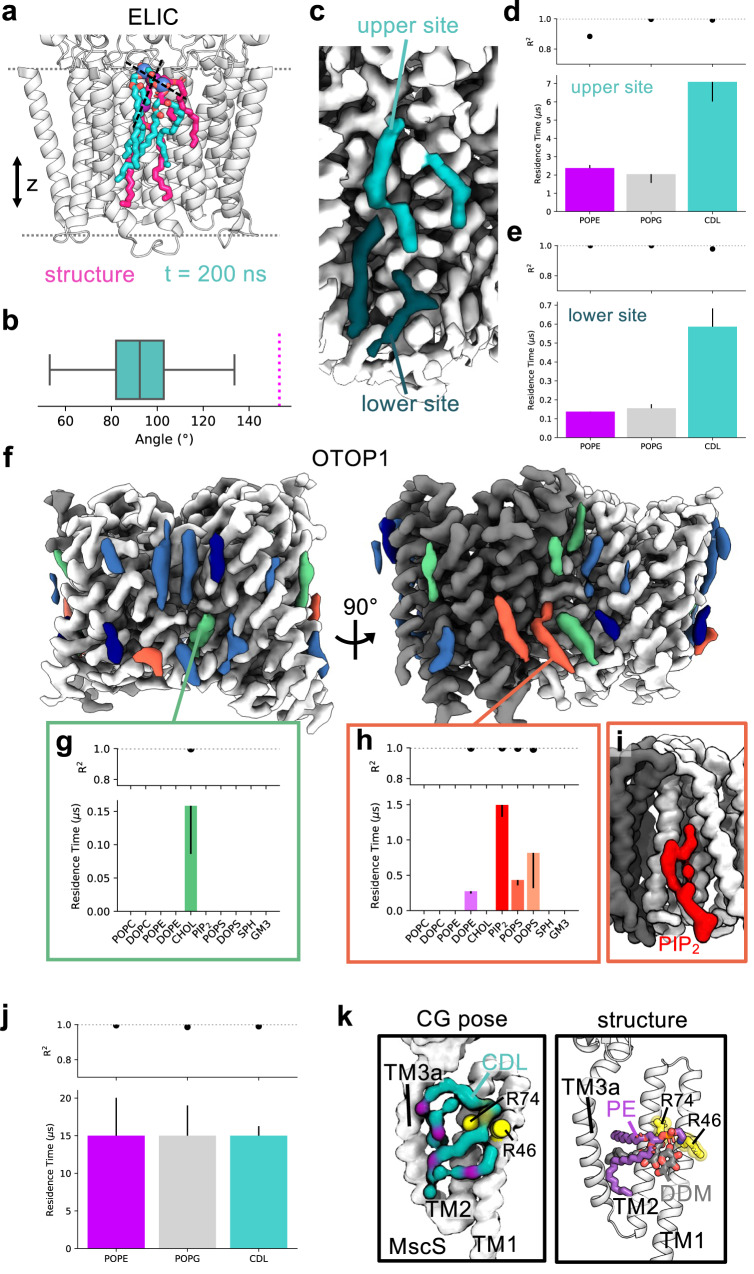
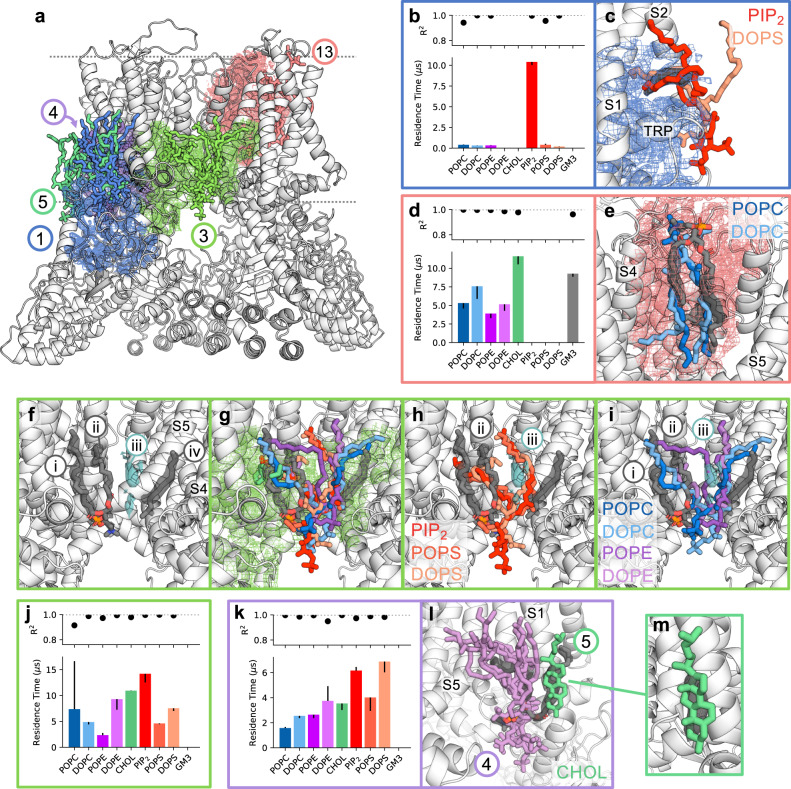
Cryo-electron microscopy (cryo-EM) enables the determination of membrane protein structures in native-like environments. Characterising how membrane proteins interact with the surrounding membrane lipid environment is assisted by resolution of lipid-like densities visible in cryo-EM maps. Nevertheless, establishing the molecular identity of putative lipid and/or detergent densities remains challenging. Here we present LipIDens, a pipeline for molecular dynamics (MD) simulation-assisted interpretation of lipid and lipid-like densities in cryo-EM structures. The pipeline integrates the implementation and analysis of multi-scale MD simulations for identification, ranking and refinement of lipid binding poses which superpose onto cryo-EM map densities. Thus, LipIDens enables direct integration of experimental and computational structural approaches to facilitate the interpretation of lipid-like cryo-EM densities and to reveal the molecular identities of protein-lipid interactions within a bilayer environment. We demonstrate this by application of our open-source LipIDens code to ten diverse membrane protein structures which exhibit lipid-like densities.
© 2023. The Author(s).
Conflict of interest statement
C.S. is a consultant for Dark Blue Therapeutics. The authors declare no other competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
