

This is a preprint.
Comprehensive Overview of Bottom-Up Proteomics using Mass Spectrometry
- PMID: 38013887
- PMCID: PMC10680866
Comprehensive Overview of Bottom-Up Proteomics using Mass Spectrometry
Update in
-
Comprehensive Overview of Bottom-Up Proteomics Using Mass Spectrometry.ACS Meas Sci Au. 2024 Jun 4;4(4):338-417. doi: 10.1021/acsmeasuresciau.3c00068. eCollection 2024 Aug 21. ACS Meas Sci Au. 2024. PMID: 39193565 Free PMC article. Review.
Abstract
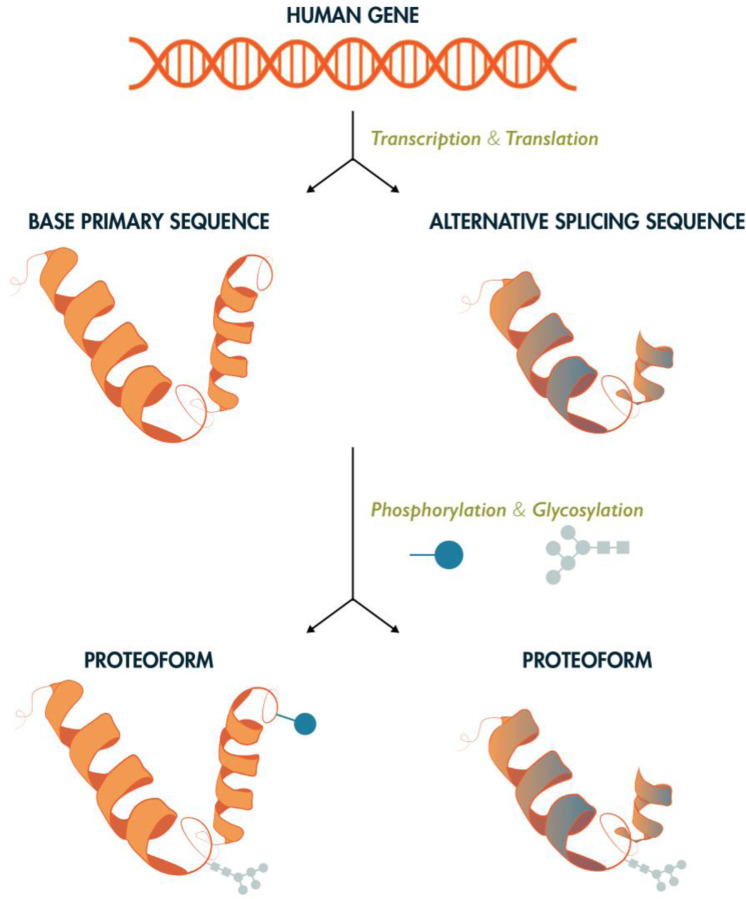
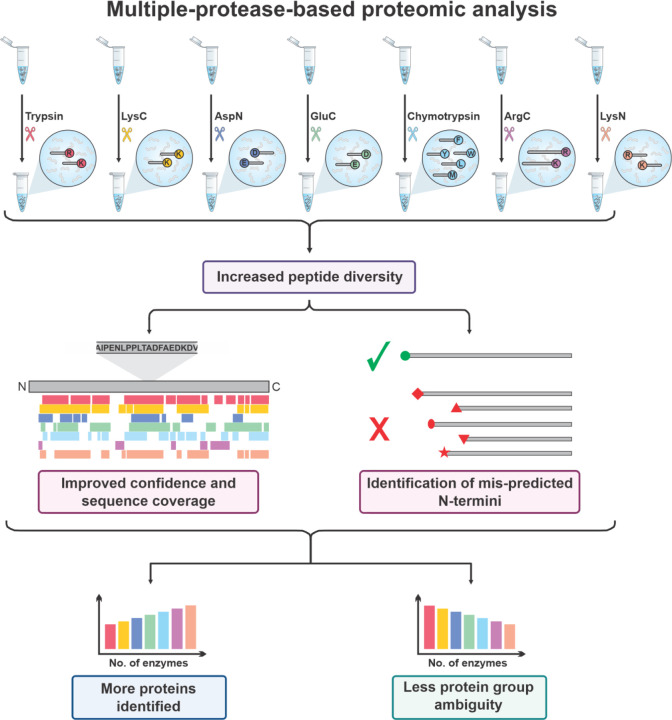
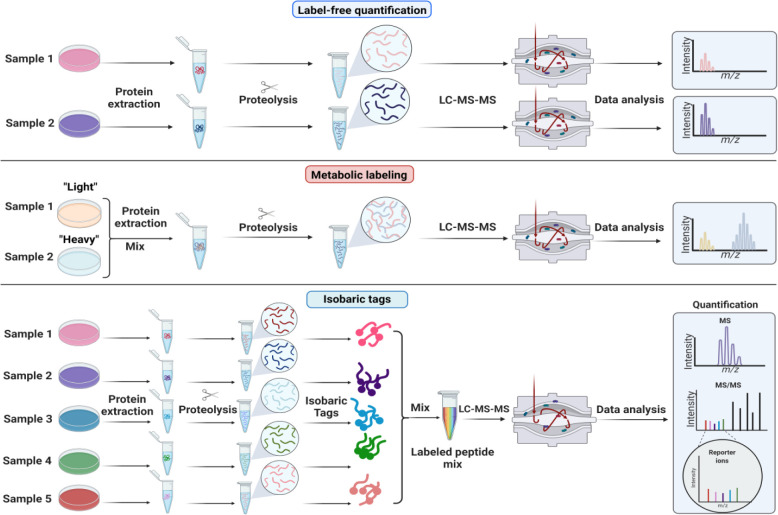
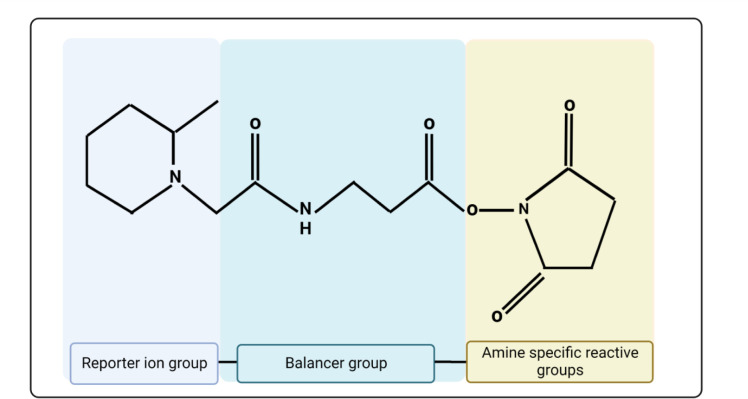
Proteomics is the large scale study of protein structure and function from biological systems through protein identification and quantification. "Shotgun proteomics" or "bottom-up proteomics" is the prevailing strategy, in which proteins are hydrolyzed into peptides that are analyzed by mass spectrometry. Proteomics studies can be applied to diverse studies ranging from simple protein identification to studies of proteoforms, protein-protein interactions, protein structural alterations, absolute and relative protein quantification, post-translational modifications, and protein stability. To enable this range of different experiments, there are diverse strategies for proteome analysis. The nuances of how proteomic workflows differ may be challenging to understand for new practitioners. Here, we provide a comprehensive overview of different proteomics methods to aid the novice and experienced researcher. We cover from biochemistry basics and protein extraction to biological interpretation and orthogonal validation. We expect this work to serve as a basic resource for new practitioners in the field of shotgun or bottom-up proteomics.
Figures
References
-
- Enzyme-less nanopore detection of post-translational modifications within long polypeptides Martin-Baniandres Pablo, Lan Wei-Hsuan, Board Stephanie, Romero-Ruiz Mercedes, Garcia-Manyes Sergi, Qing Yujia, Bayley Hagan Nature Nanotechnology (2023-July-27) https://doi.org/gs3f47 DOI: 10.1038/s41565-023-01462-8 - DOI - PMC - PubMed
-
- Nanopore Detection Using Supercharged Polypeptide Molecular Carriers Wang Xiaoyi, Thomas Tina-Marie, Ren Ren, Zhou Yu, Zhang Peng, Li Jingjing, Cai Shenglin, Liu Kai, Ivanov Aleksandar P, Herrmann Andreas, Edel Joshua B Journal of the American Chemical Society (2023-March-10) https://doi.org/gs3f46 DOI: 10.1021/jacs.2c13465 - DOI - PMC - PubMed
-
- Real-time shape approximation and fingerprinting of single proteins using a nanopore Yusko Erik C, Bruhn Brandon R, Eggenberger Olivia M, Houghtaling Jared, Rollings Ryan C, Walsh Nathan C, Nandivada Santoshi, Pindrus Mariya, Hall Adam R, Sept David, … Mayer Michael Nature Nanotechnology (2016-December-19) https://doi.org/gfx2jk DOI: 10.1038/nnano.2016.267 - DOI - PubMed
-
- Nanopore-Based Protein Identification Bakshloo Mazdak Afshar, Kasianowicz John J, Pastoriza-Gallego Manuela, Mathé Jérôme, Daniel Régis, Piguet Fabien, Oukhaled Abdelghani Journal of the American Chemical Society (2022-February-04) https://doi.org/gr9grc DOI: 10.1021/jacs.1c11758 - DOI - PubMed
-
- Highly parallel single-molecule identification of proteins in zeptomole-scale mixtures Swaminathan Jagannath, Boulgakov Alexander A, Hernandez Erik T, Bardo Angela M, Bachman James L, Marotta Joseph, Johnson Amber M, Anslyn Eric V, Marcotte Edward M Nature Biotechnology (2018-October-22) https://doi.org/gfg4bk DOI: 10.1038/nbt.4278 - DOI - PMC - PubMed