

An evolutionary perspective on complex neuropsychiatric disease
- PMID: 38016473
- PMCID: PMC10842497
- DOI: 10.1016/j.neuron.2023.10.037
An evolutionary perspective on complex neuropsychiatric disease
Abstract
The forces of evolution-mutation, selection, migration, and genetic drift-shape the genetic architecture of human traits, including the genetic architecture of complex neuropsychiatric illnesses. Studying these illnesses in populations that are diverse in genetic ancestry, historical demography, and cultural history can reveal how evolutionary forces have guided adaptation over time and place. A fundamental truth of shared human biology is that an allele responsible for a disease in anyone, anywhere, reveals a gene critical to the normal biology underlying that condition in everyone, everywhere. Understanding the genetic causes of neuropsychiatric disease in the widest possible range of human populations thus yields the greatest possible range of insight into genes critical to human brain development. In this perspective, we explore some of the relationships between genes, adaptation, and history that can be illuminated by an evolutionary perspective on studies of complex neuropsychiatric disease in diverse populations.
Keywords: 22q11 deletion; OCD; assortative mating; autism; bipolar disorder; causality; clinical heterogeneity; complex neuropsychiatric disease; consanguinity; de novo mutation; evolution; genetic drift; genetics; genomics; migration; polygenic inheritance; rare alleles; schizophrenia; selection; somatic mutation.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests D.S. has received consultancy honoraria from Discovery Vitality, Johnson & Johnson, Kanna, L’Oreal, Lundbeck, Orion, Sanofi, Servier, Takeda, and Vistagen.
Figures




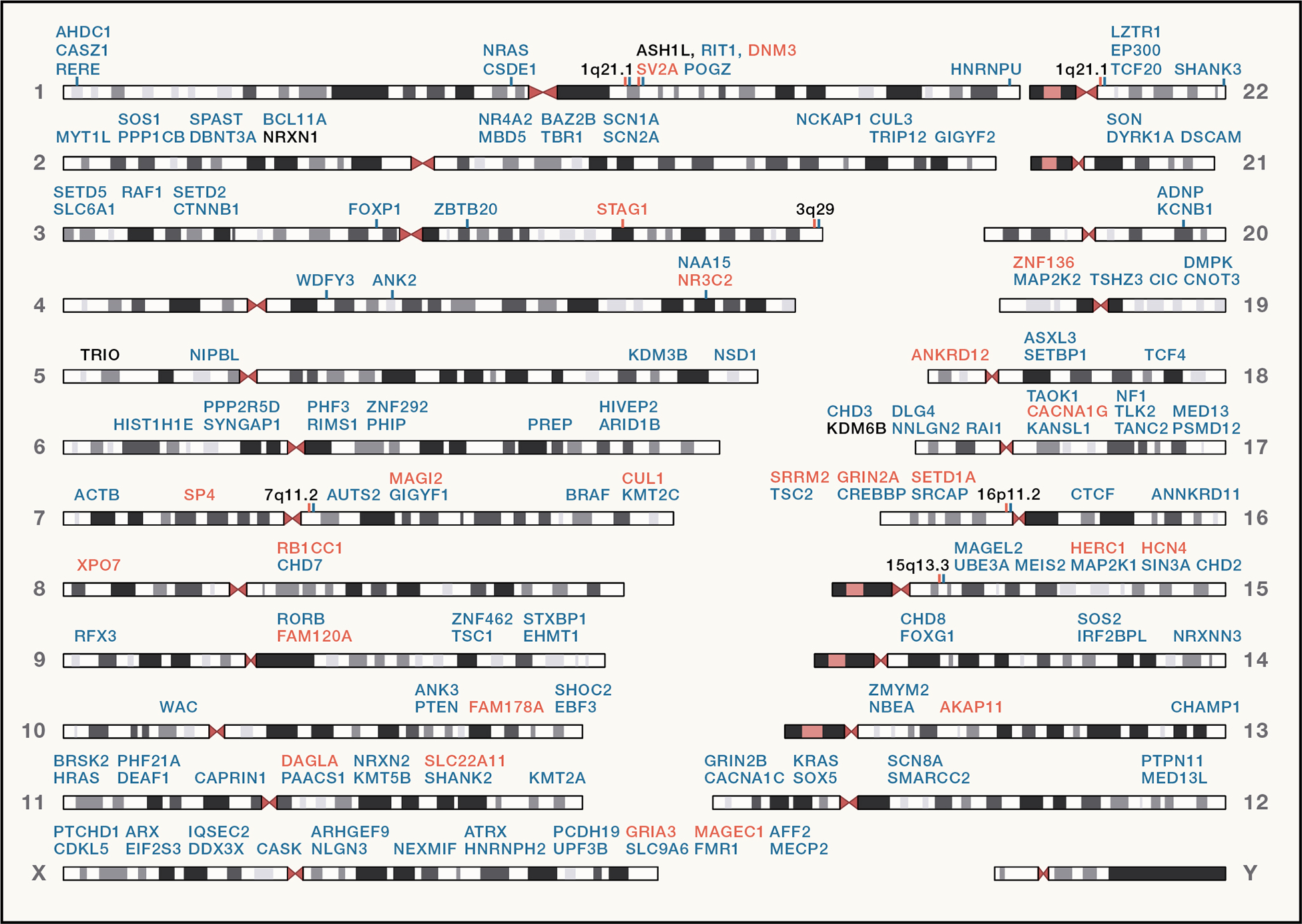

References
-
- Dobzhansky T (1973). Nothing in Biology Makes Sense Except in the Light of Evolution. Am. Biol. Teach. 35, 125–129. 10.2307/4444260. - DOI
Publication types
MeSH terms
Grants and funding
- K99 MH128540/MH/NIMH NIH HHS/United States
- U01 MH125053/MH/NIMH NIH HHS/United States
- R01 MH124679/MH/NIMH NIH HHS/United States
- U01 MH125062/MH/NIMH NIH HHS/United States
- U01 MH126798/MH/NIMH NIH HHS/United States
- U01 MH125054/MH/NIMH NIH HHS/United States
- R01 MH123451/MH/NIMH NIH HHS/United States
- R01 MH130675/MH/NIMH NIH HHS/United States
- P50 HD105351/HD/NICHD NIH HHS/United States
- R01 MH130674/MH/NIMH NIH HHS/United States
- R01 MH128813/MH/NIMH NIH HHS/United States
- U01 MH125058/MH/NIMH NIH HHS/United States
- U01 MH124962/MH/NIMH NIH HHS/United States
- U01 MH125050/MH/NIMH NIH HHS/United States
