

Dynamics of water-mediated interaction effects on the stability and transmission of Omicron
- PMID: 38017052
- PMCID: PMC10684572
- DOI: 10.1038/s41598-023-48186-2
Dynamics of water-mediated interaction effects on the stability and transmission of Omicron
Abstract
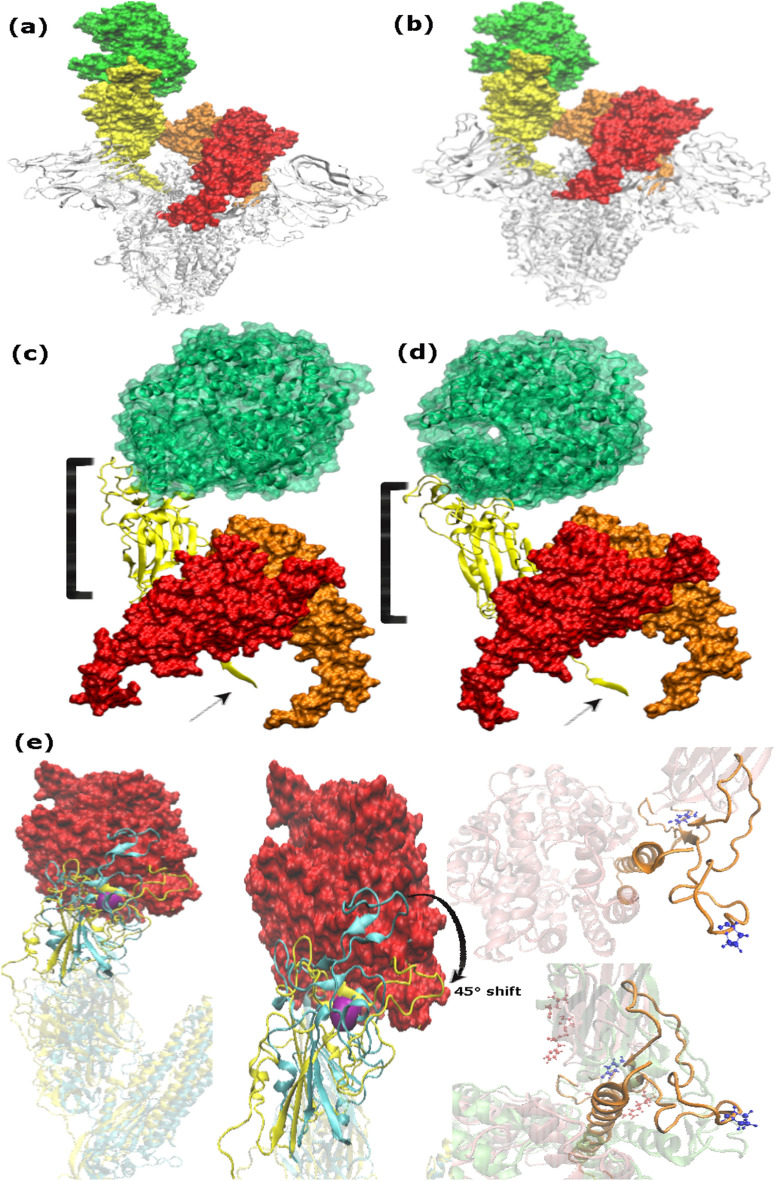
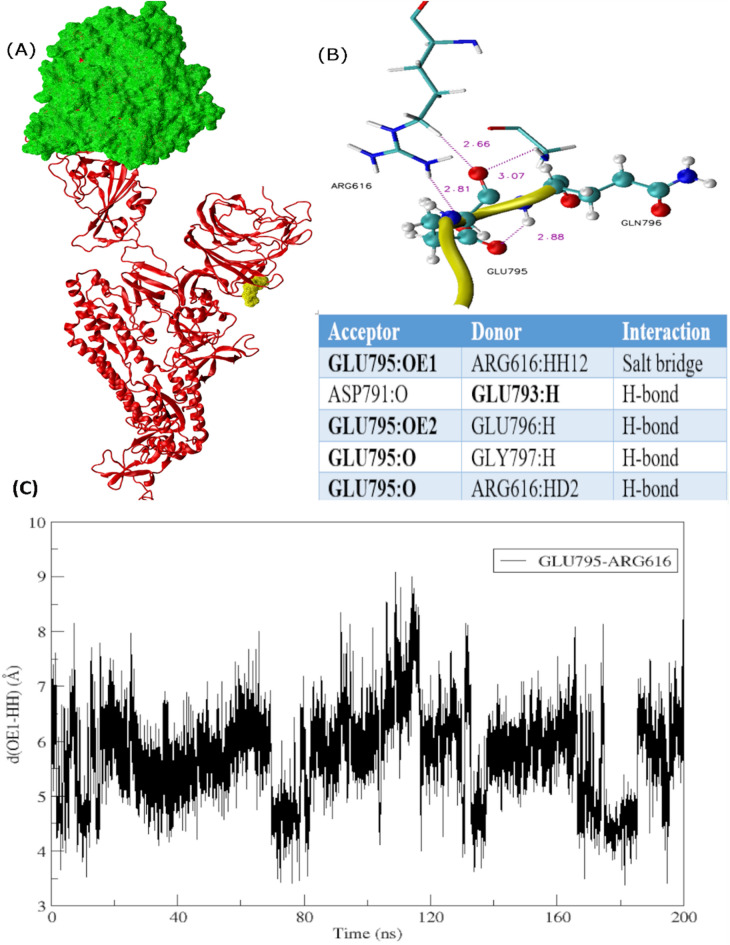
SARS-Cov-2 Omicron variant and its highly transmissible sublineages amidst news of emerging hybrid variants strengthen the evidence of its ability to rapidly spread and evolve giving rise to unprecedented future waves. Owing to the presence of isolated RBD, monomeric and trimeric Cryo-EM structures of spike protein in complex with ACE2 receptor, comparative analysis of Alpha, Beta, Gamma, Delta, and Omicron assist in a rational assessment of their probability to evolve as new or hybrid variants in future. This study proposes the role of hydration forces in mediating Omicron function and dynamics based on a stronger interplay between protein and solvent with each Covid wave. Mutations of multiple hydrophobic residues into hydrophilic residues underwent concerted interactions with water leading to variations in charge distribution in Delta and Omicron during molecular dynamics simulations. Moreover, comparative analysis of interacting moieties characterized a large number of mutations lying at RBD into constrained, homologous and low-affinity groups referred to as mutational drivers inferring that the probability of future mutations relies on their function. Furthermore, the computational findings reveal a significant difference in angular distances among variants of concern due 3 amino acid insertion (EPE) in Omicron variant that not only facilitates tight domain organization but also seems requisite for characterization of mutational processes. The outcome of this work signifies the possible relation between hydration forces, their impact on conformation and binding affinities, and viral fitness that will significantly aid in understanding dynamics of drug targets for Covid-19 countermeasures. The emerging scenario is that hydration forces and hydrophobic interactions are crucial variables to probe in mutational analysis to explore conformational landscape of macromolecules and reveal the molecular origins of protein behaviors.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures






References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
