

Assessing protein model quality based on deep graph coupled networks using protein language model
- PMID: 38018909
- PMCID: PMC10685403
- DOI: 10.1093/bib/bbad420
Assessing protein model quality based on deep graph coupled networks using protein language model
Abstract
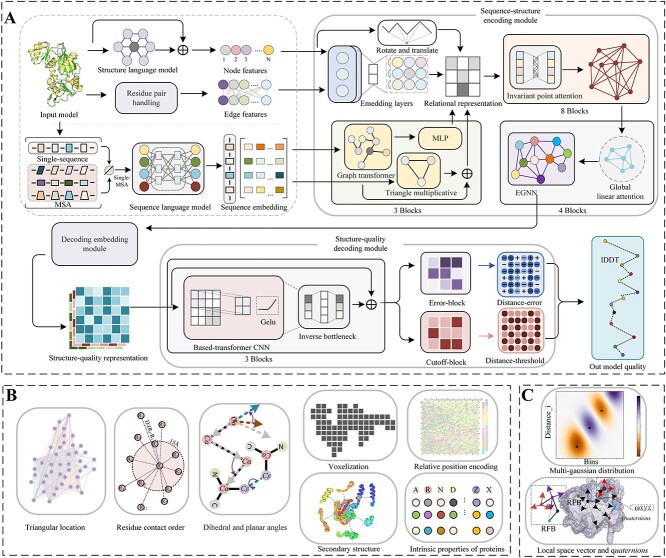
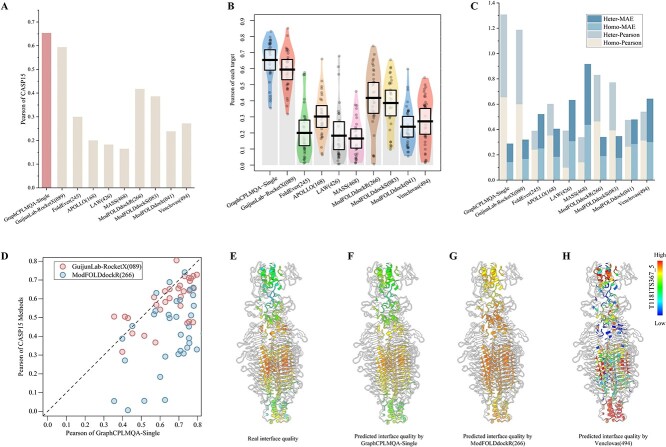
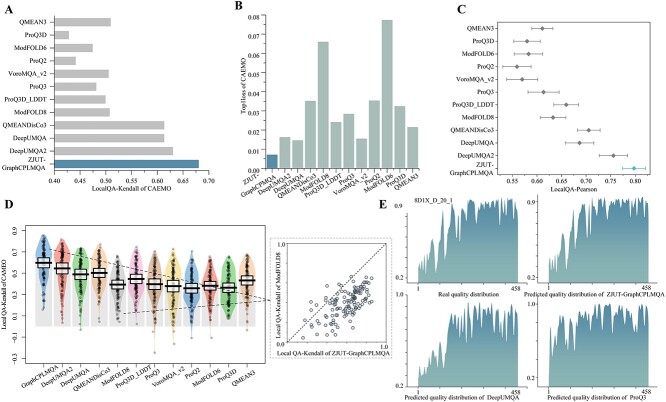
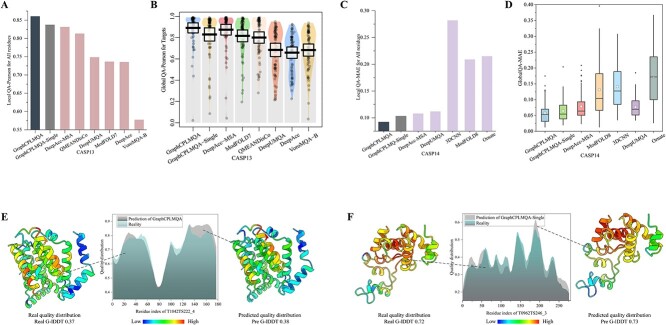
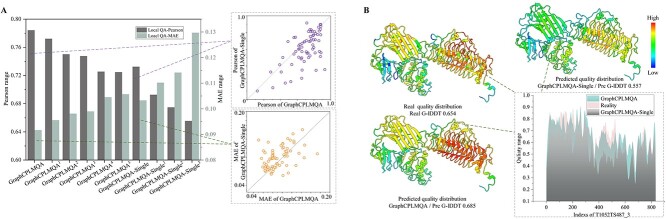
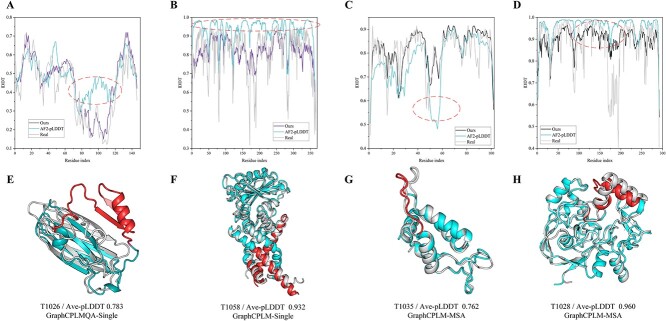
Model quality evaluation is a crucial part of protein structural biology. How to distinguish high-quality models from low-quality models, and to assess which high-quality models have relatively incorrect regions for improvement, are remain a challenge. More importantly, the quality assessment of multimer models is a hot topic for structure prediction. In this study, we propose GraphCPLMQA, a novel approach for evaluating residue-level model quality that combines graph coupled networks and embeddings from protein language models. The GraphCPLMQA consists of a graph encoding module and a transform-based convolutional decoding module. In encoding module, the underlying relational representations of sequence and high-dimensional geometry structure are extracted by protein language models with Evolutionary Scale Modeling. In decoding module, the mapping connection between structure and quality is inferred by the representations and low-dimensional features. Specifically, the triangular location and residue level contact order features are designed to enhance the association between the local structure and the overall topology. Experimental results demonstrate that GraphCPLMQA using single-sequence embedding achieves the best performance compared with the CASP15 residue-level interface evaluation methods among 9108 models in the local residue interface test set of CASP15 multimers. In CAMEO blind test (20 May 2022 to 13 August 2022), GraphCPLMQA ranked first compared with other servers (https://www.cameo3d.org/quality-estimation). GraphCPLMQA also outperforms state-of-the-art methods on 19, 035 models in CASP13 and CASP14 monomer test set.
Keywords: graph neural network; multimer model evaluation; protein language model; protein model evaluation.
© The Author(s) 2023. Published by Oxford University Press.
Figures
References
-
- Lin Z, Akin H, Rao R, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023;379:1123–1130. - PubMed
