

Enhanced TP53 reactivation disrupts MYC transcriptional program and overcomes venetoclax resistance in acute myeloid leukemias
- PMID: 38019903
- PMCID: PMC10686564
- DOI: 10.1126/sciadv.adh1436
Enhanced TP53 reactivation disrupts MYC transcriptional program and overcomes venetoclax resistance in acute myeloid leukemias
Abstract
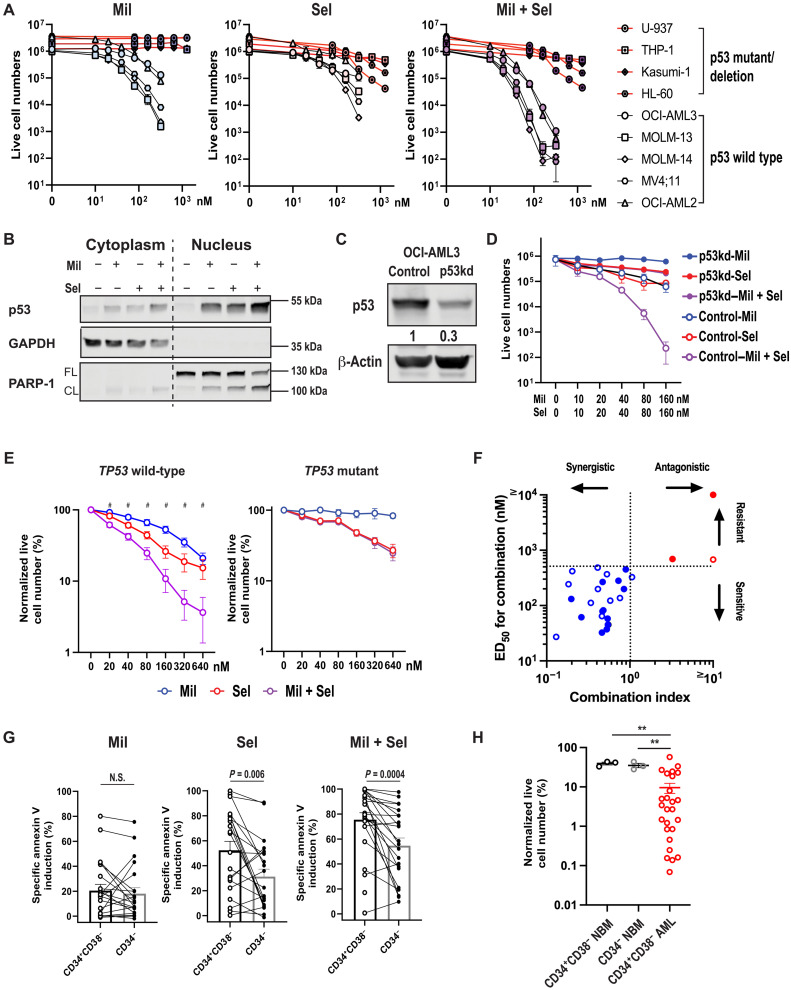
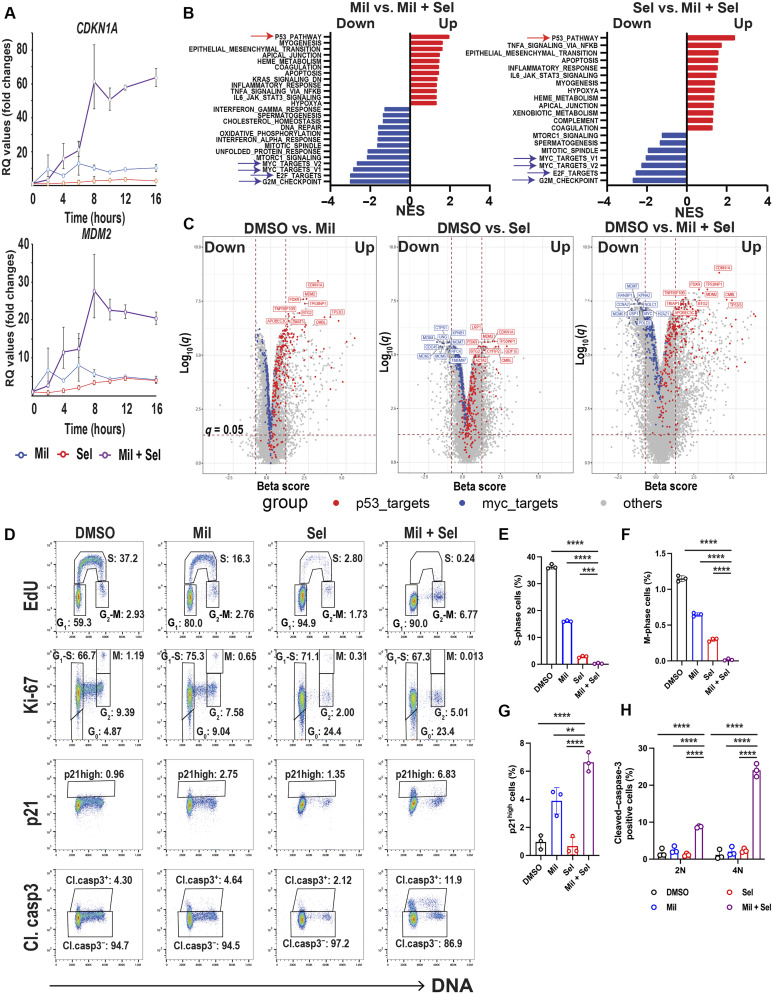
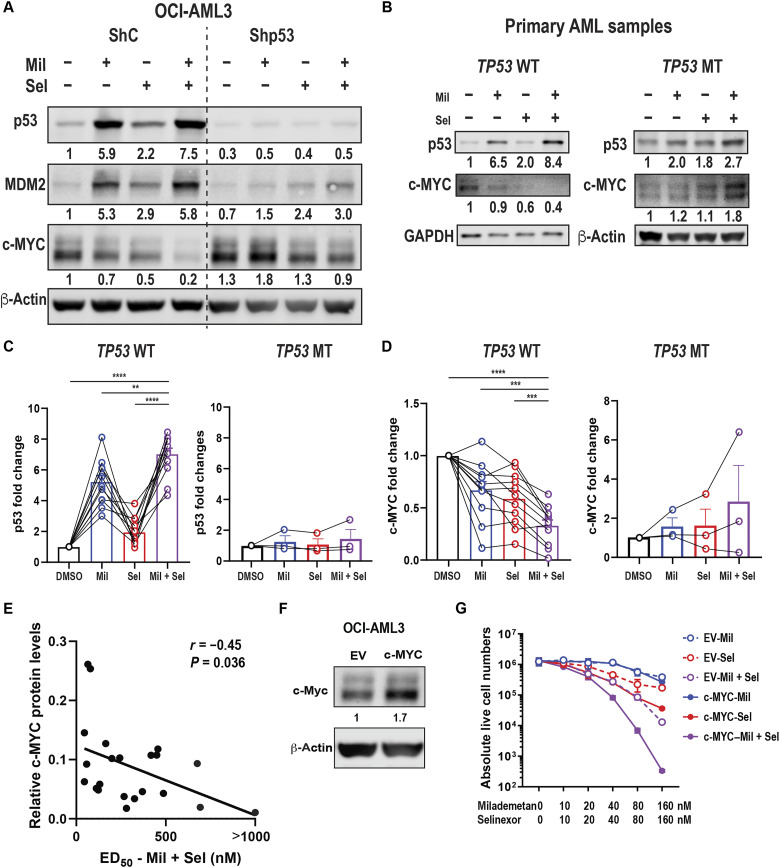
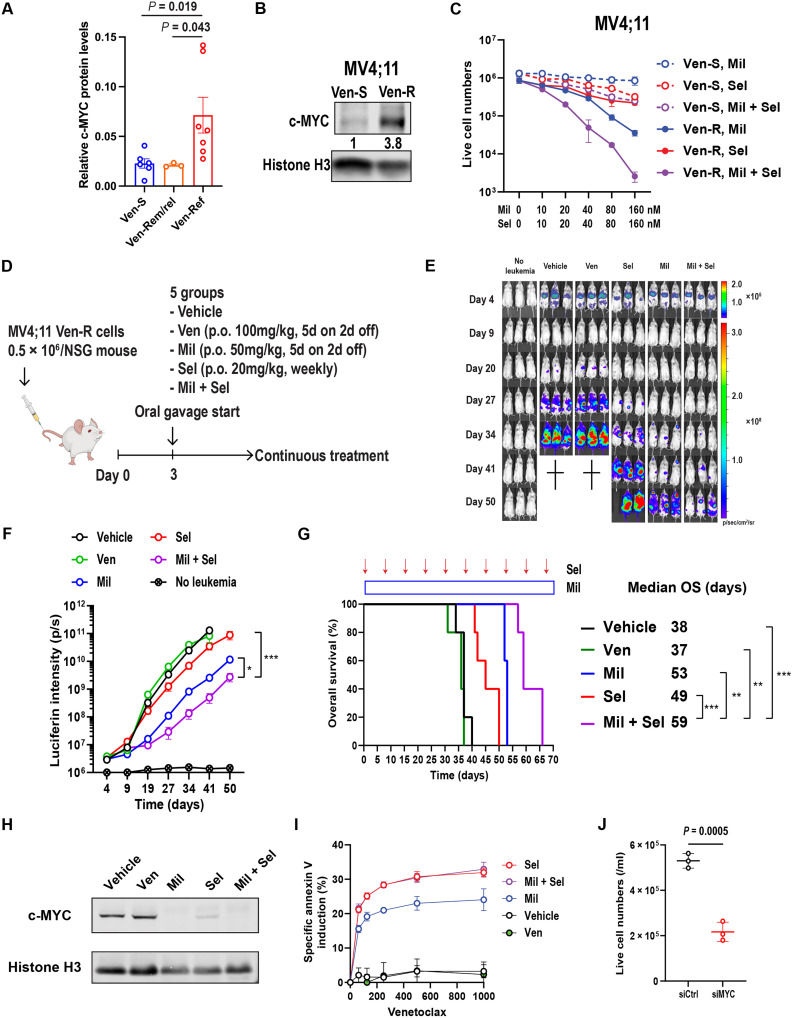
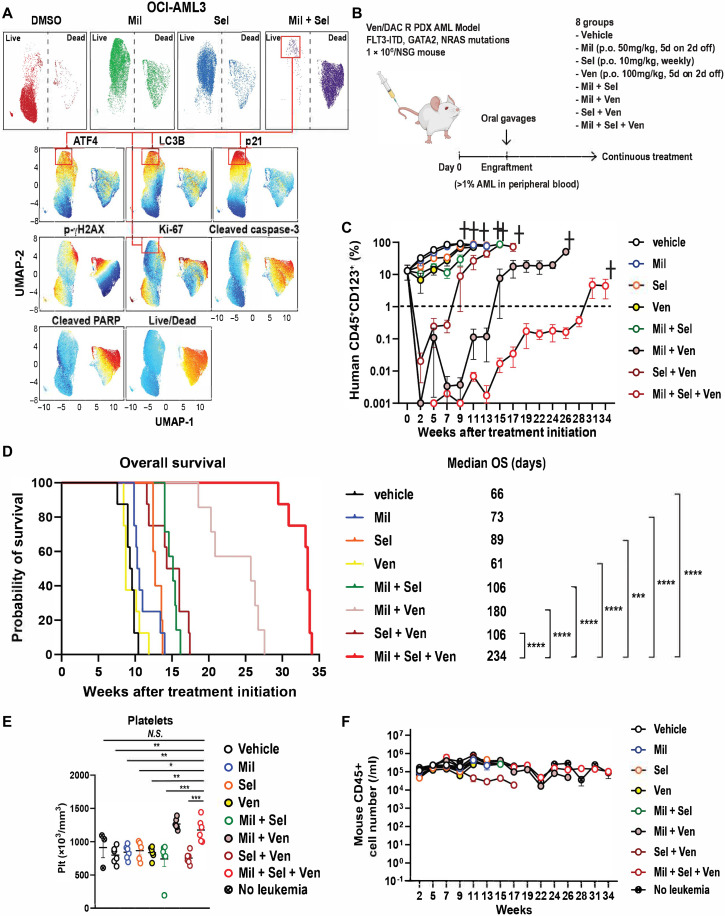
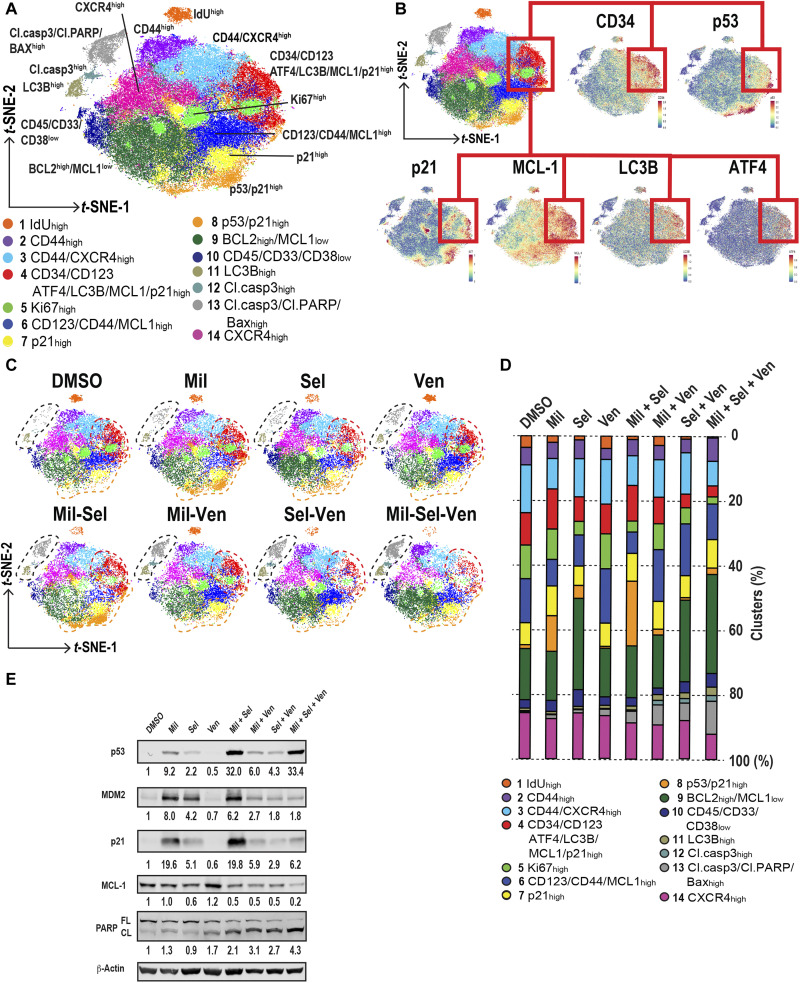
The tumor suppressor TP53 is frequently inactivated in a mutation-independent manner in cancers and is reactivated by inhibiting its negative regulators. We here cotarget MDM2 and the nuclear exporter XPO1 to maximize transcriptional activity of p53. MDM2/XPO1 inhibition accumulated nuclear p53 and elicited a 25- to 60-fold increase of its transcriptional targets. TP53 regulates MYC, and MDM2/XPO1 inhibition disrupted the c-MYC-regulated transcriptome, resulting in the synergistic induction of apoptosis in acute myeloid leukemia (AML). Unexpectedly, venetoclax-resistant AMLs express high levels of c-MYC and are vulnerable to MDM2/XPO1 inhibition in vivo. However, AML cells persisting after MDM2/XPO1 inhibition exhibit a quiescence- and stress response-associated phenotype. Venetoclax overcomes that resistance, as shown by single-cell mass cytometry. The triple inhibition of MDM2, XPO1, and BCL2 was highly effective against venetoclax-resistant AML in vivo. Our results propose a novel, highly translatable therapeutic approach leveraging p53 reactivation to overcome nongenetic, stress-adapted venetoclax resistance.
Figures
References
-
- A. Vazquez, E. E. Bond, A. J. Levine, G. L. Bond, The genetics of the p53 pathway, apoptosis and cancer therapy. Nat. Rev. Drug Discov. 7, 979–987 (2008). - PubMed
-
- T. J. Phesse, K. B. Myant, A. M. Cole, R. A. Ridgway, H. Pearson, V. Muncan, G. R. van den Brink, K. H. Vousden, R. Sears, L. T. Vassilev, A. R. Clarke, O. J. Sansom, Endogenous c-Myc is essential for p53-induced apoptosis in response to DNA damage in vivo. Cell Death Differ. 21, 956–966 (2014). - PMC - PubMed
-
- C. Lourenco, D. Resetca, C. Redel, P. Lin, A. S. MacDonald, R. Ciaccio, T. M. G. Kenney, Y. Wei, D. W. Andrews, M. Sunnerhagen, C. H. Arrowsmith, B. Raught, L. Z. Penn, MYC protein interactors in gene transcription and cancer. Nat. Rev. Cancer 21, 579–591 (2021). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
