

Identification of a regulatory pathway governing TRAF1 via an arthritis-associated non-coding variant
- PMID: 38020975
- PMCID: PMC10667332
- DOI: 10.1016/j.xgen.2023.100420
Identification of a regulatory pathway governing TRAF1 via an arthritis-associated non-coding variant
Erratum in
-
Identification of a regulatory pathway governing TRAF1 via an arthritis-associated non-coding variant.Cell Genom. 2024 Feb 14;4(2):100502. doi: 10.1016/j.xgen.2024.100502. Cell Genom. 2024. PMID: 38359789 Free PMC article. No abstract available.
Abstract
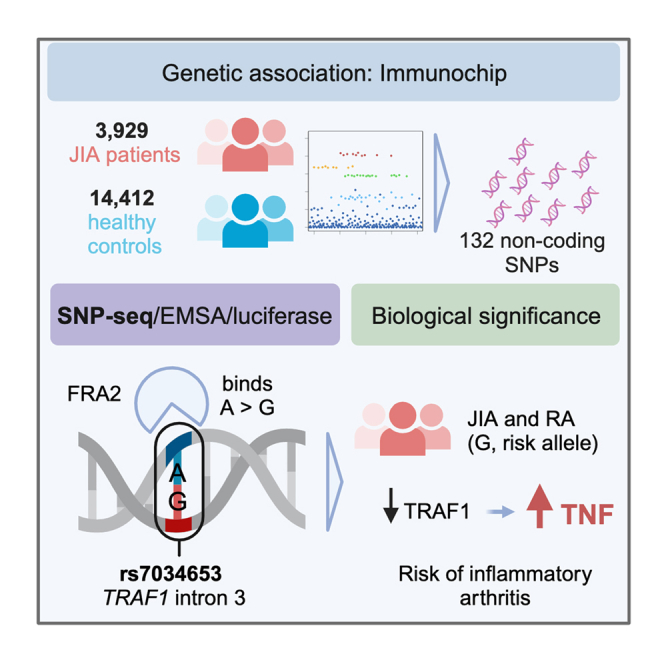
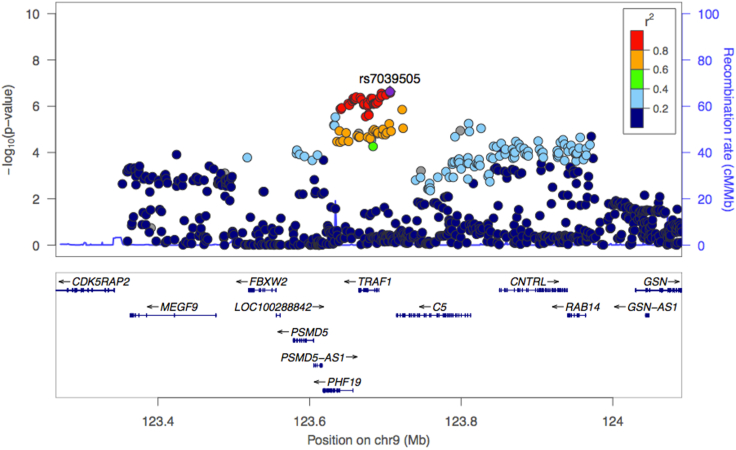
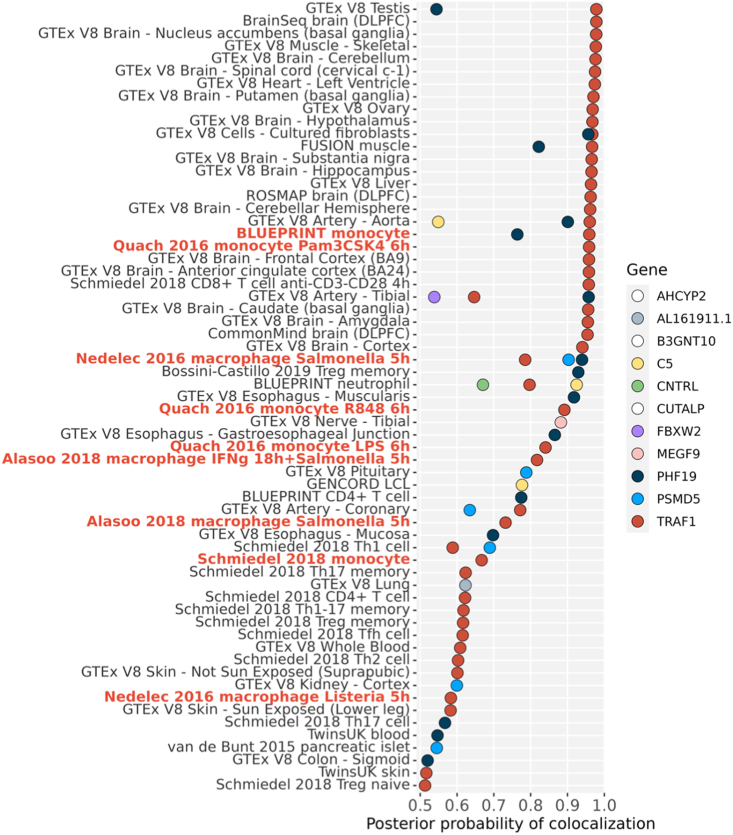
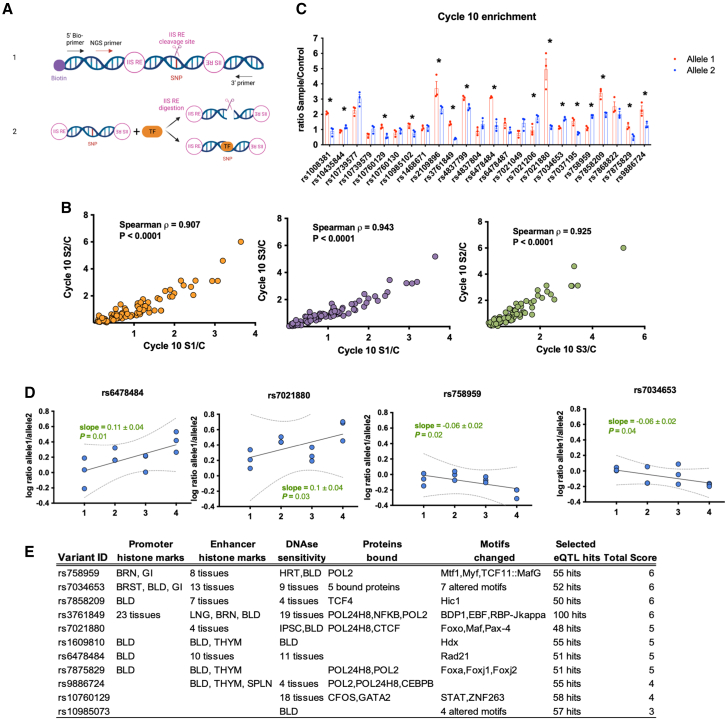
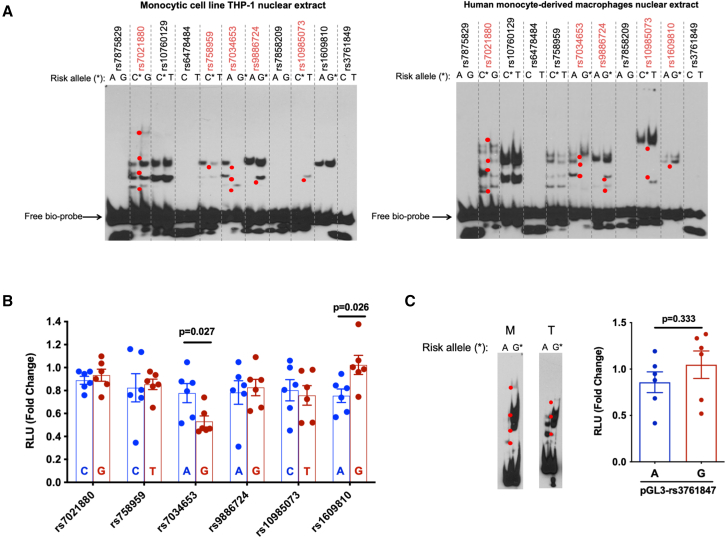
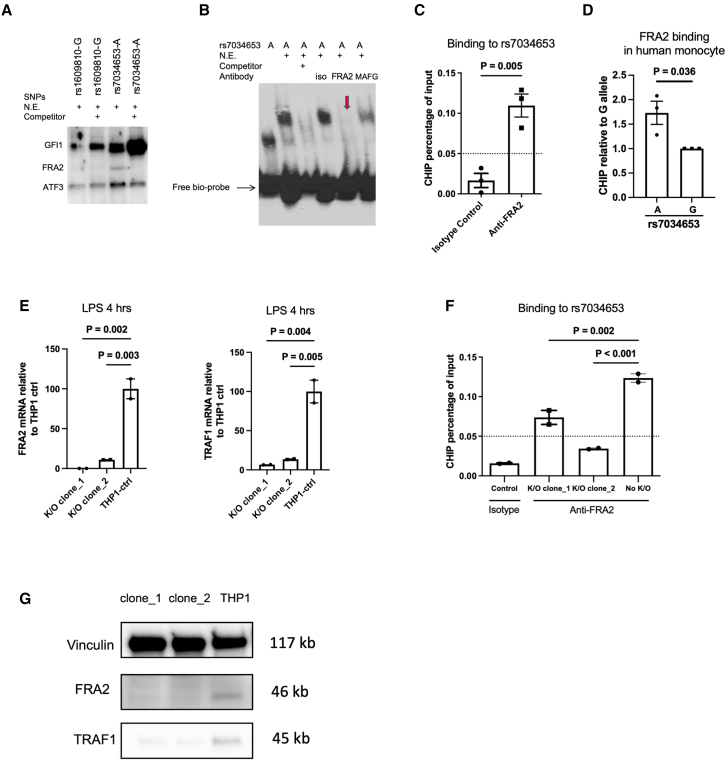
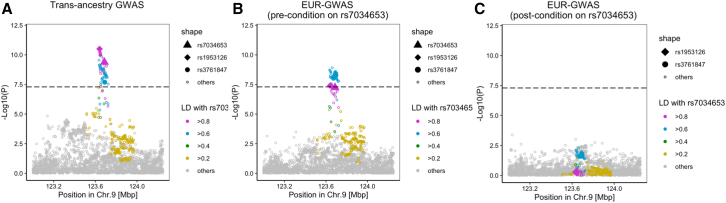
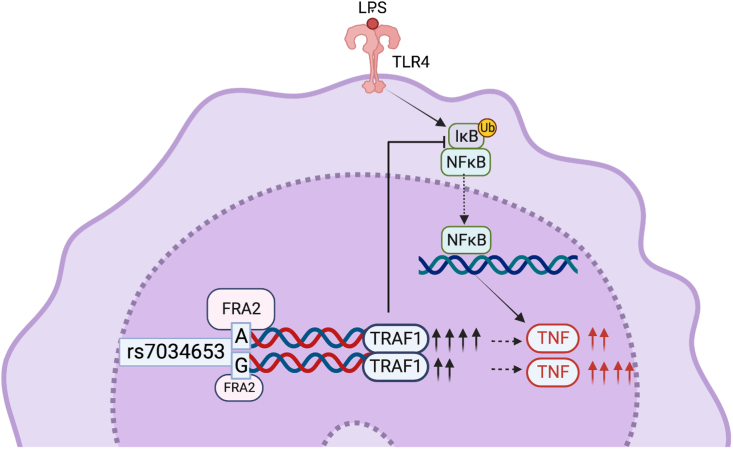
TRAF1/C5 was among the first loci shown to confer risk for inflammatory arthritis in the absence of an associated coding variant, but its genetic mechanism remains undefined. Using Immunochip data from 3,939 patients with juvenile idiopathic arthritis (JIA) and 14,412 control individuals, we identified 132 plausible common non-coding variants, reduced serially by single-nucleotide polymorphism sequencing (SNP-seq), electrophoretic mobility shift, and luciferase studies to the single variant rs7034653 in the third intron of TRAF1. Genetically manipulated experimental cells and primary monocytes from genotyped donors establish that the risk G allele reduces binding of Fos-related antigen 2 (FRA2), encoded by FOSL2, resulting in reduced TRAF1 expression and enhanced tumor necrosis factor (TNF) production. Conditioning on this JIA variant eliminated attributable risk for rheumatoid arthritis, implicating a mechanism shared across the arthritis spectrum. These findings reveal that rs7034653, FRA2, and TRAF1 mediate a pathway through which a non-coding functional variant drives risk of inflammatory arthritis in children and adults.
Keywords: C5; FRA2; TNF; TRAF1; genome-wide association study; juvenile idiopathic arthritis; monocyte; non-coding variant; rheumatoid arthritis.
© 2023 The Authors.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Grants and funding
- U01 AI130830/AI/NIAID NIH HHS/United States
- R01 AR073228/AR/NIAMS NIH HHS/United States
- R01 NS099068/NS/NINDS NIH HHS/United States
- P30 AR072577/AR/NIAMS NIH HHS/United States
- R01 AR073201/AR/NIAMS NIH HHS/United States
- P30 AR070253/AR/NIAMS NIH HHS/United States
- R01 AR065538/AR/NIAMS NIH HHS/United States
- R01 AI024717/AI/NIAID NIH HHS/United States
- P30 AR070549/AR/NIAMS NIH HHS/United States
- R01 AR063759/AR/NIAMS NIH HHS/United States
- R01 AR075906/AR/NIAMS NIH HHS/United States
- P30 AR069625/AR/NIAMS NIH HHS/United States
- R01 AR077607/AR/NIAMS NIH HHS/United States
- R01 HD089928/HD/NICHD NIH HHS/United States
- R01 HG010730/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous
