

Organic Selenium induces ferroptosis in pancreatic cancer cells
- PMID: 38029455
- PMCID: PMC10698006
- DOI: 10.1016/j.redox.2023.102962
Organic Selenium induces ferroptosis in pancreatic cancer cells
Abstract
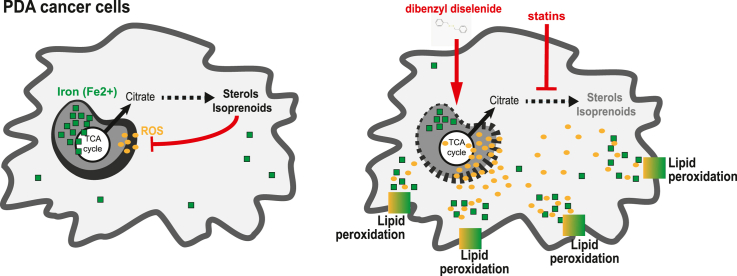
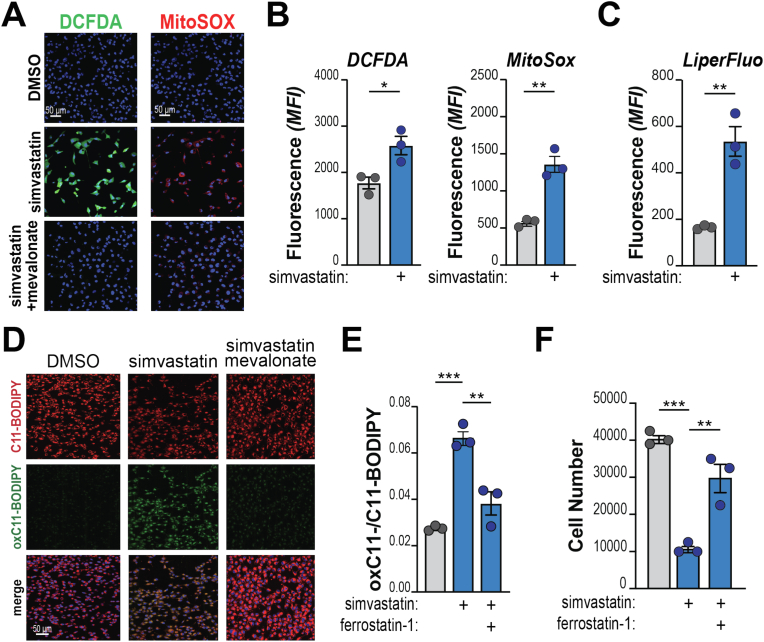
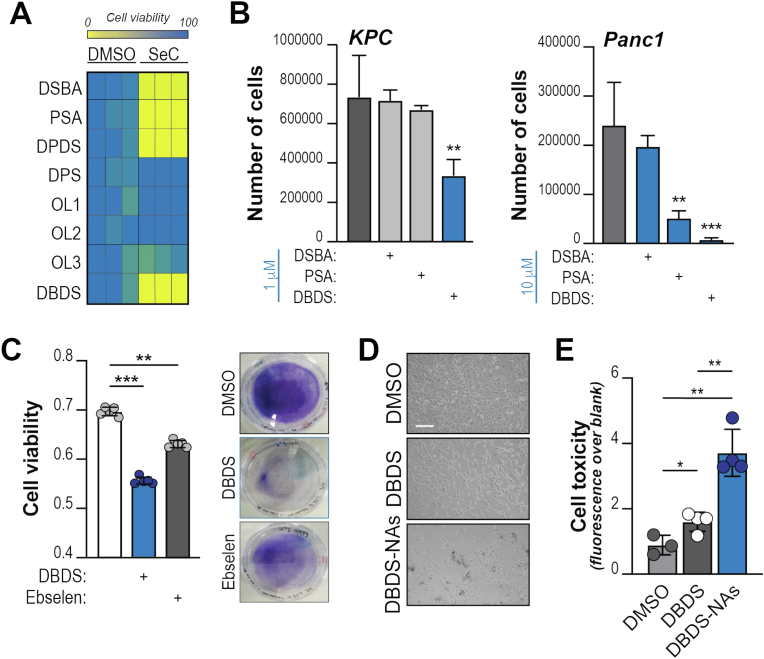
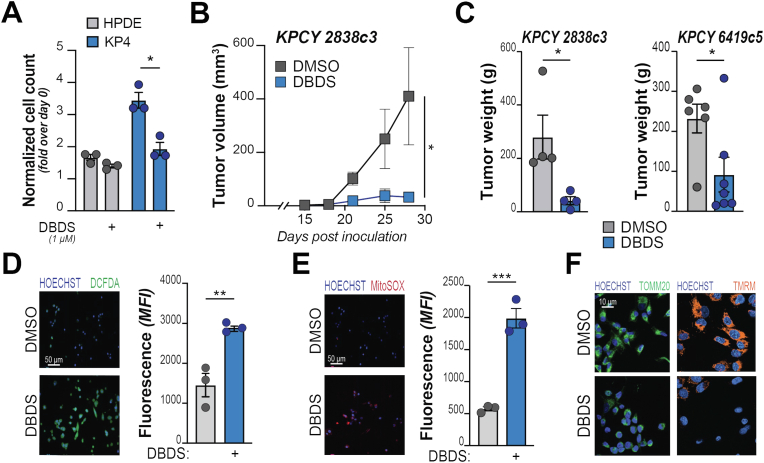
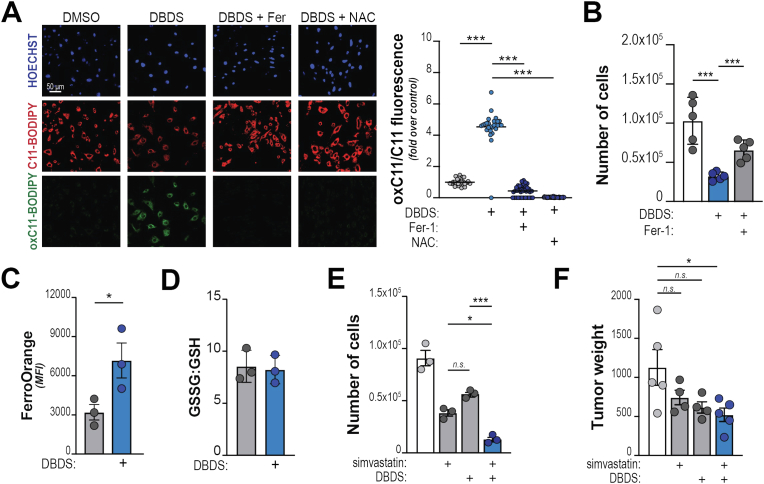
Pancreatic ductal adenocarcinoma (PDA) cells reprogram both mitochondrial and lysosomal functions to support growth. At the same time, this causes significant dishomeostasis of free radicals. While this is compensated by the upregulation of detoxification mechanisms, it also represents a potential vulnerability. Here we demonstrate that PDA cells are sensitive to the inhibition of the mevalonate pathway (MVP), which supports the biosynthesis of critical antioxidant intermediates and protect from ferroptosis. We attacked the susceptibility of PDA cells to ferroptotic death with selenorganic compounds, including dibenzyl diselenide (DBDS) that exhibits potent pro-oxidant properties and inhibits tumor growth in vitro and in vivo. DBDS treatment induces the mobilization of iron from mitochondria enabling uncontrolled lipid peroxidation. Finally, we showed that DBDS and statins act synergistically to promote ferroptosis and provide evidence that combined treatment is a viable strategy to combat PDA.
Keywords: Dibenzyl diselenide (DBDS); Ferroptosis; Mevalonate pathway (MVP); Pancreatic cancer; Selenorganic compounds.
Copyright © 2023 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of competing interest None.
Figures
References
-
- He D., Feng H., Sundberg B., Yang J., Powers J., Christian A.H., Wilkinson J.E., Monnin C., Avizonis D., Thomas C.J., Friedman R.A., Kluger M.D., Hollingsworth M.A., Grandgenett P.M., Klute K.A., Toste F.D., Chang C.J., Chio I.I.C. Methionine oxidation activates pyruvate kinase M2 to promote pancreatic cancer metastasis. Mol. Cell. 2022;82:3045–3060. doi: 10.1016/j.molcel.2022.06.005. e11. - DOI - PMC - PubMed
-
- Bar-Peled L., Kemper E.K., Suciu R.M., V Vinogradova E., Backus K.M., Horning B.D., Paul T.A., Ichu T.-A., Svensson R.U., Olucha J., Chang M.W., Kok B.P., Zhu Z., Ihle N.T., Dix M.M., Jiang P., Hayward M.M., Saez E., Shaw R.J., Cravatt B.F. Chemical proteomics identifies druggable vulnerabilities in a genetically defined cancer. Cell. 2017;171:696–709.e23. doi: 10.1016/j.cell.2017.08.051. - DOI - PMC - PubMed
-
- Nieborowska-Skorska M., Kopinski P.K., Ray R., Hoser G., Ngaba D., Flis S., Cramer K., Reddy M.M., Koptyra M., Penserga T., Glodkowska-Mrowka E., Bolton E., Holyoake T.L., Eaves C.J., Cerny-Reiterer S., Valent P., Hochhaus A., Hughes T.P., van der Kuip H., Sattler M., Wiktor-Jedrzejczak W., Richardson C., Dorrance A., Stoklosa T., Williams D.A., Skorski T. Rac2-MRC-cIII–generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood. 2012;119:4253–4263. doi: 10.1182/blood-2011-10-385658. - DOI - PMC - PubMed
-
- Romani P., Nirchio N., Arboit M., Barbieri V., Tosi A., Michielin F., Shibuya S., Benoist T., Wu D., Hindmarch C.C.T., Giomo M., Urciuolo A., Giamogante F., Roveri A., Chakravarty P., Montagner M., Calì T., Elvassore N., Archer S.L., De Coppi P., Rosato A., Martello G., Dupont S. Mitochondrial fission links ECM mechanotransduction to metabolic redox homeostasis and metastatic chemotherapy resistance. Nat. Cell Biol. 2022;24:168–180. doi: 10.1038/s41556-022-00843-w. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
