

A rapid, low-cost, and highly sensitive SARS-CoV-2 diagnostic based on whole-genome sequencing
- PMID: 38032990
- PMCID: PMC10688730
- DOI: 10.1371/journal.pone.0294283
A rapid, low-cost, and highly sensitive SARS-CoV-2 diagnostic based on whole-genome sequencing
Abstract
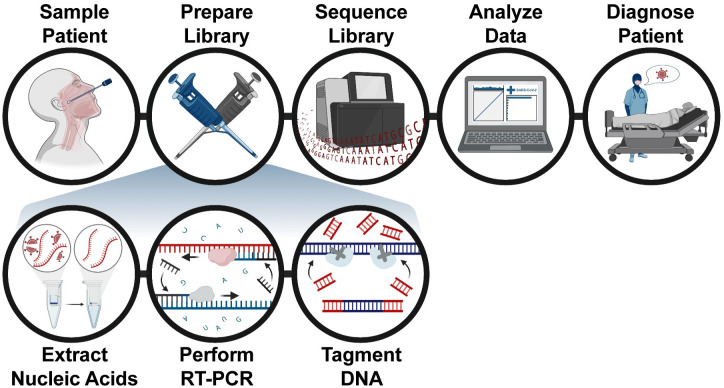
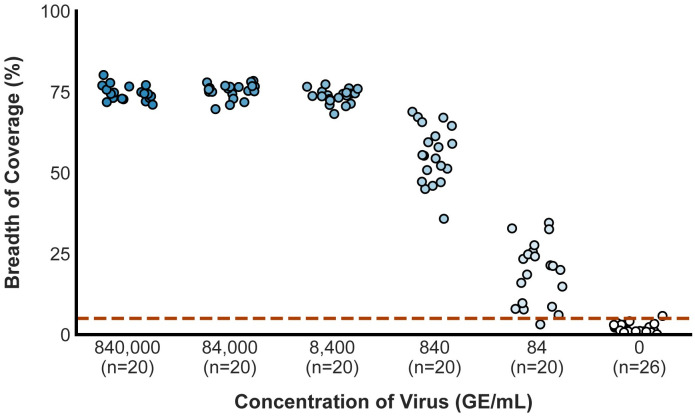
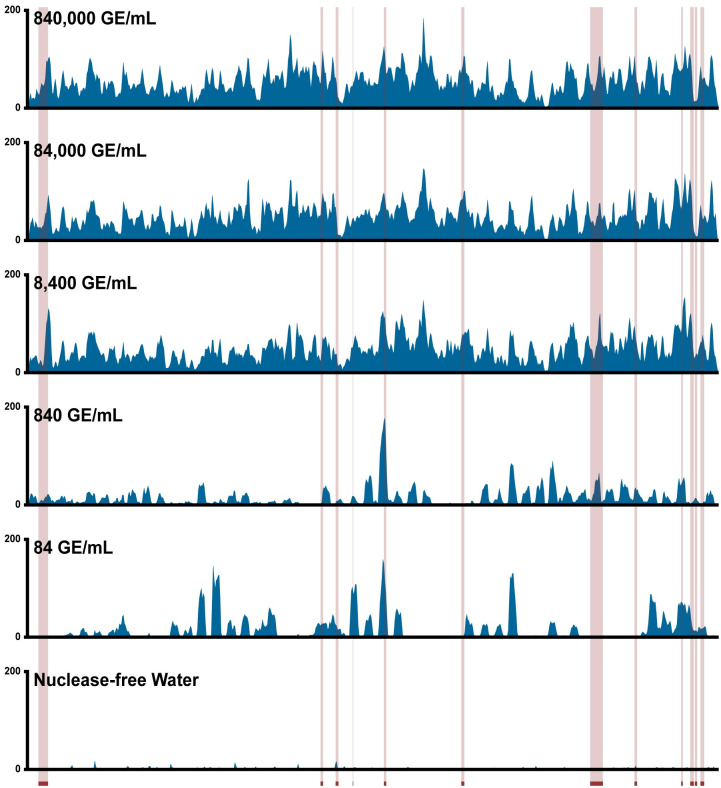
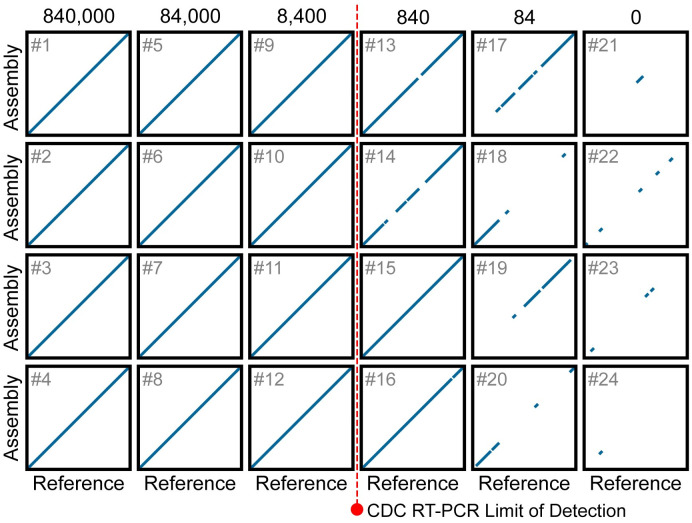
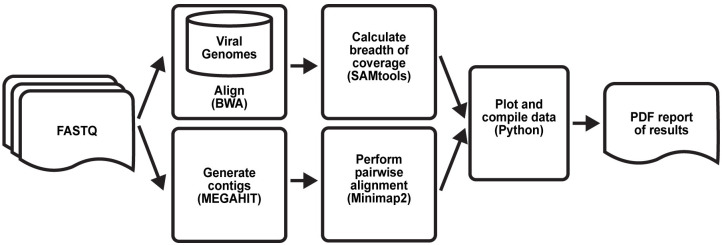
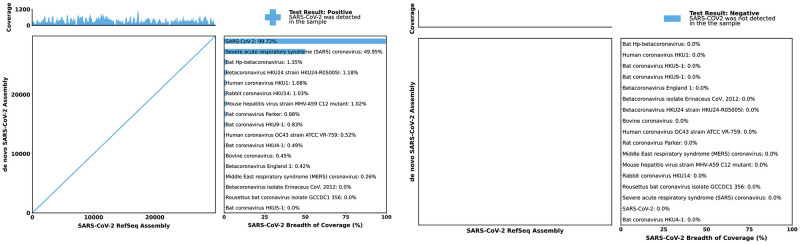
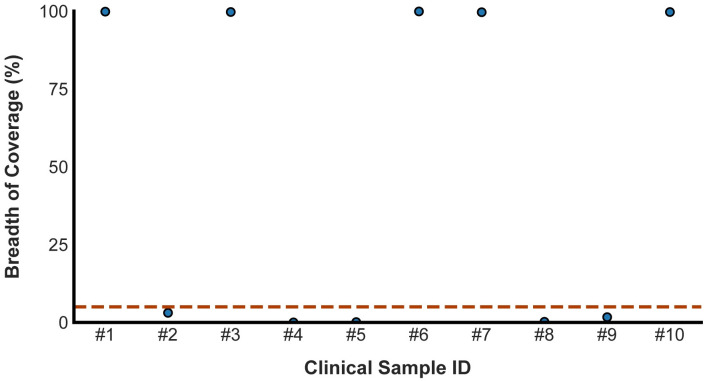
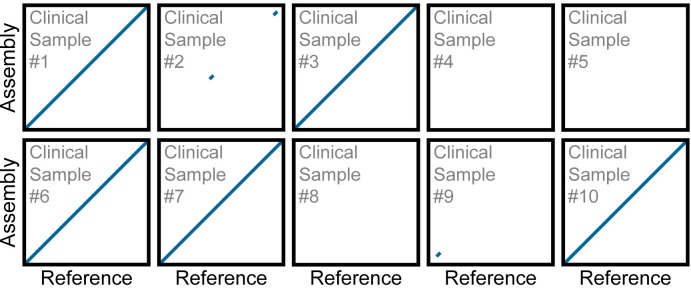
Early detection of SARS-CoV-2 infection is key to managing the current global pandemic, as evidence shows the virus is most contagious on or before symptom onset. Here, we introduce a low-cost, high-throughput method for diagnosing and studying SARS-CoV-2 infection. Dubbed Pathogen-Oriented Low-Cost Assembly & Re-Sequencing (POLAR), this method amplifies the entirety of the SARS-CoV-2 genome. This contrasts with typical RT-PCR-based diagnostic tests, which amplify only a few loci. To achieve this goal, we combine a SARS-CoV-2 enrichment method developed by the ARTIC Network (https://artic.network/) with short-read DNA sequencing and de novo genome assembly. Using this method, we can reliably (>95% accuracy) detect SARS-CoV-2 at a concentration of 84 genome equivalents per milliliter (GE/mL). The vast majority of diagnostic methods meeting our analytical criteria that are currently authorized for use by the United States Food and Drug Administration with the Coronavirus Disease 2019 (COVID-19) Emergency Use Authorization require higher concentrations of the virus to achieve this degree of sensitivity and specificity. In addition, we can reliably assemble the SARS-CoV-2 genome in the sample, often with no gaps and perfect accuracy given sufficient viral load. The genotypic data in these genome assemblies enable the more effective analysis of disease spread than is possible with an ordinary binary diagnostic. These data can also help identify vaccine and drug targets. Finally, we show that the diagnoses obtained using POLAR of positive and negative clinical nasal mid-turbinate swab samples 100% match those obtained in a clinical diagnostic lab using the Center for Disease Control's 2019-Novel Coronavirus test. Using POLAR, a single person can manually process 192 samples over an 8-hour experiment at the cost of ~$36 per patient (as of December 7th, 2022), enabling a 24-hour turnaround with sequencing and data analysis time. We anticipate that further testing and refinement will allow greater sensitivity using this approach.
Copyright: © 2023 Adastra et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
-
- Worldometers.info. COVID Live—Coronavirus Statistics—Worldometer. 9 Nov 2022 [cited 7 Nov 2022]. https://www.worldometers.info/coronavirus/.
-
- To KKW, Tsang OTY, Leung WS, Tam AR, Wu TC, Lung DC, et al.. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: an observational cohort study. Lancet Infect Dis. 2020;20: 565–574. doi: 10.1016/S1473-3099(20)30196-1 - DOI - PMC - PubMed
-
- SARS-CoV-2 Viral Mutations: Impact on COVID-19 Tests | FDA. [cited 8 Dec 2022]. https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-dev....
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
