

Frontiers of molecular crystal structure prediction for pharmaceuticals and functional organic materials
- PMID: 38033897
- PMCID: PMC10685338
- DOI: 10.1039/d3sc03903j
Frontiers of molecular crystal structure prediction for pharmaceuticals and functional organic materials
Abstract
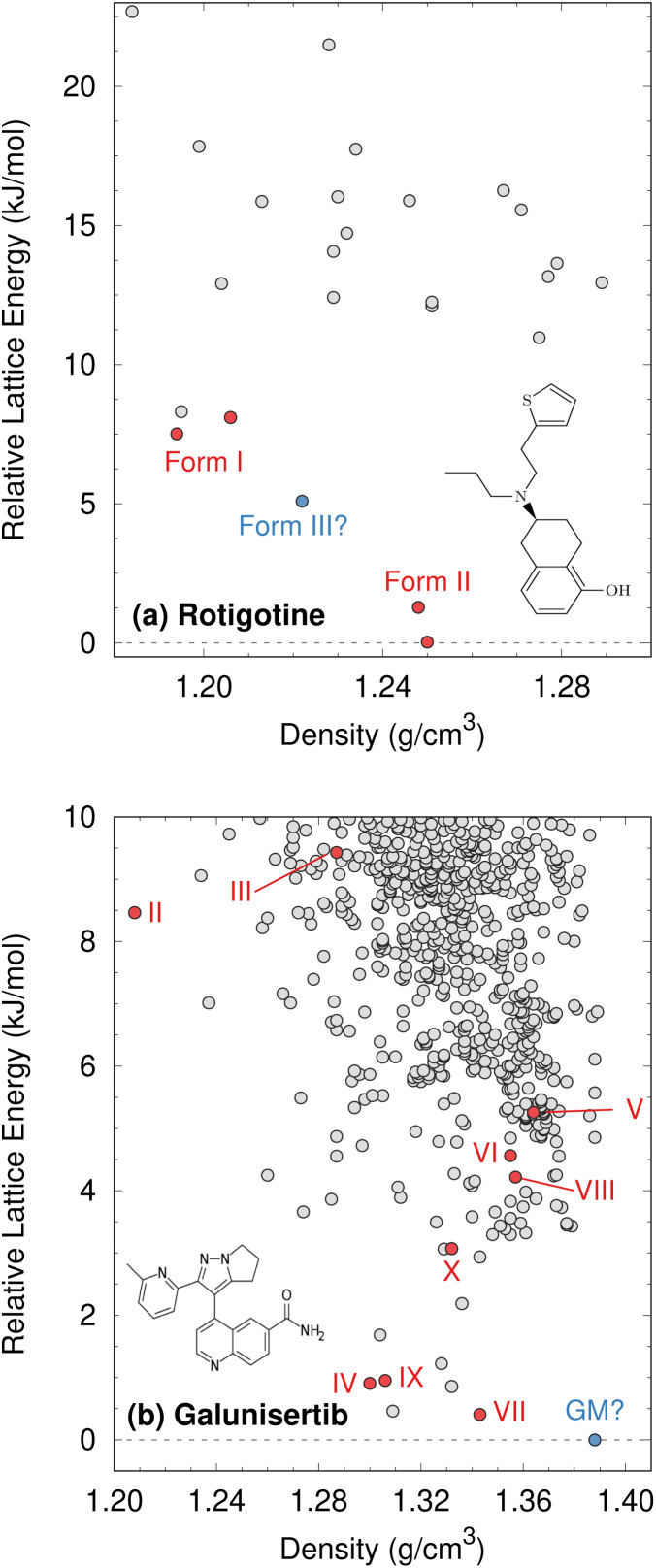
The reliability of organic molecular crystal structure prediction has improved tremendously in recent years. Crystal structure predictions for small, mostly rigid molecules are quickly becoming routine. Structure predictions for larger, highly flexible molecules are more challenging, but their crystal structures can also now be predicted with increasing rates of success. These advances are ushering in a new era where crystal structure prediction drives the experimental discovery of new solid forms. After briefly discussing the computational methods that enable successful crystal structure prediction, this perspective presents case studies from the literature that demonstrate how state-of-the-art crystal structure prediction can transform how scientists approach problems involving the organic solid state. Applications to pharmaceuticals, porous organic materials, photomechanical crystals, organic semi-conductors, and nuclear magnetic resonance crystallography are included. Finally, efforts to improve our understanding of which predicted crystal structures can actually be produced experimentally and other outstanding challenges are discussed.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
There are no conflicts to declare.
Figures













References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources