

CaMKK2 and CHK1 phosphorylate human STN1 in response to replication stress to protect stalled forks from aberrant resection
- PMID: 38036565
- PMCID: PMC10689503
- DOI: 10.1038/s41467-023-43685-2
CaMKK2 and CHK1 phosphorylate human STN1 in response to replication stress to protect stalled forks from aberrant resection
Abstract
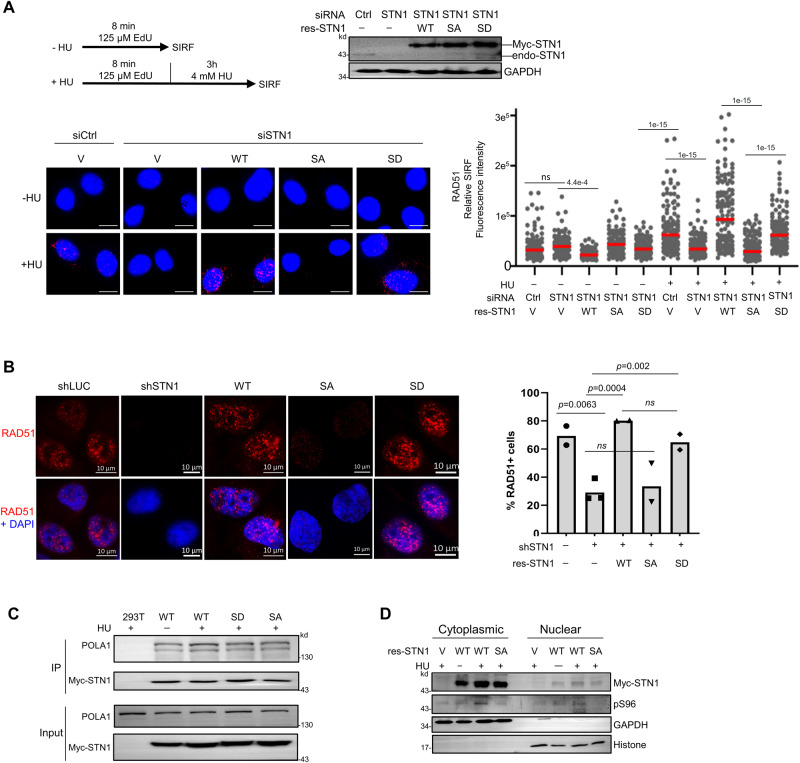
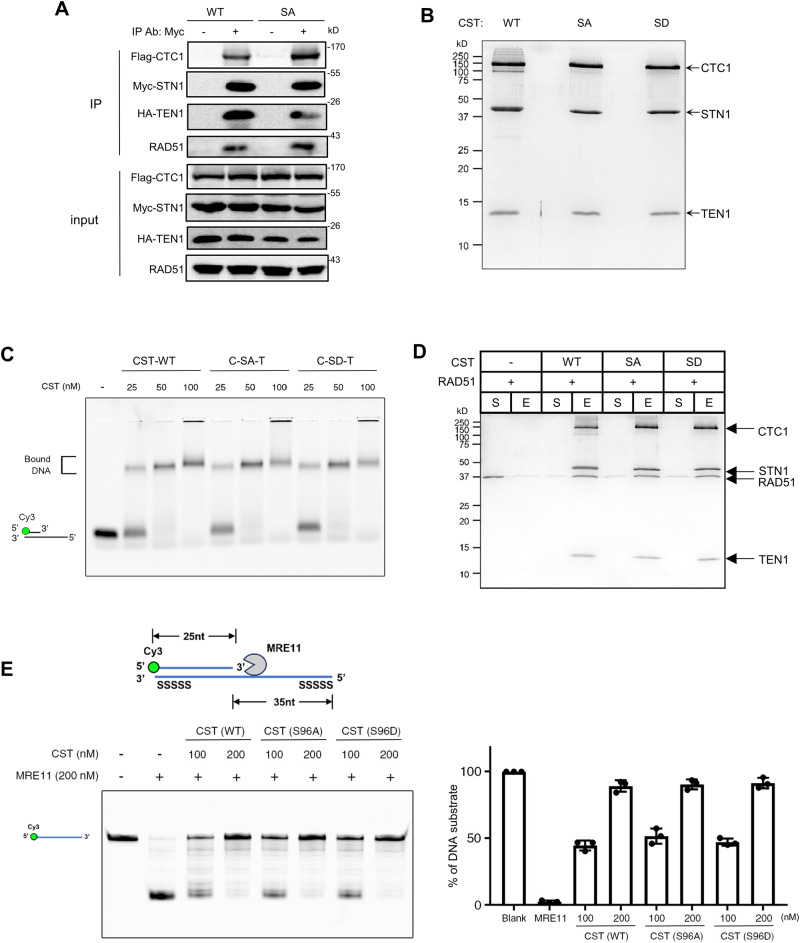
Keeping replication fork stable is essential for safeguarding genome integrity; hence, its protection is highly regulated. The CTC1-STN1-TEN1 (CST) complex protects stalled forks from aberrant MRE11-mediated nascent strand DNA degradation (NSD). However, the activation mechanism for CST at forks is unknown. Here, we report that STN1 is phosphorylated in its intrinsic disordered region. Loss of STN1 phosphorylation reduces the replication stress-induced STN1 localization to stalled forks, elevates NSD, increases MRE11 access to stalled forks, and decreases RAD51 localization at forks, leading to increased genome instability under perturbed DNA replication condition. STN1 is phosphorylated by both the ATR-CHK1 and the calcium-sensing kinase CaMKK2 in response to hydroxyurea/aphidicolin treatment or elevated cytosolic calcium concentration. Cancer-associated STN1 variants impair STN1 phosphorylation, conferring inability of fork protection. Collectively, our study uncovers that CaMKK2 and ATR-CHK1 target STN1 to enable its fork protective function, and suggests an important role of STN1 phosphorylation in cancer development.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
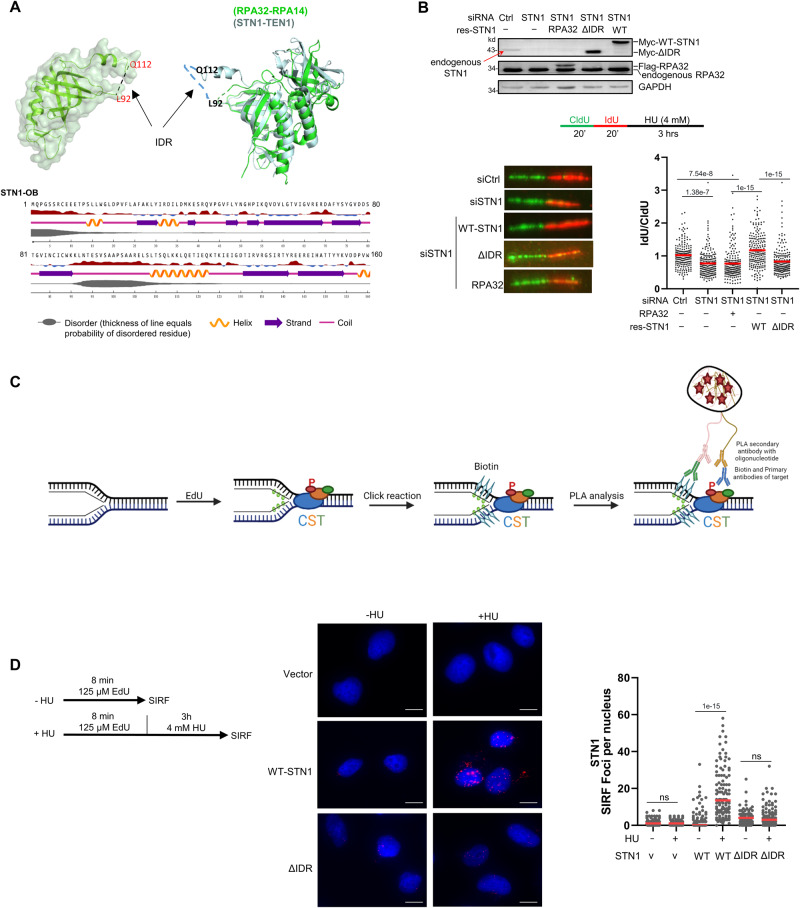
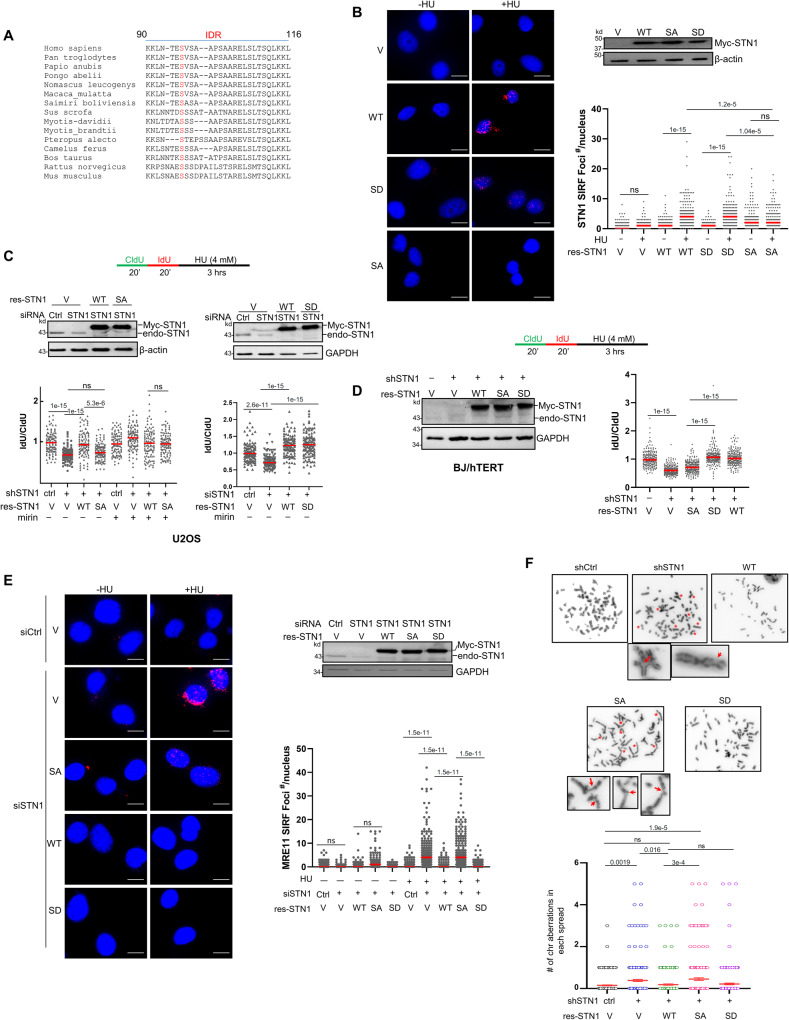
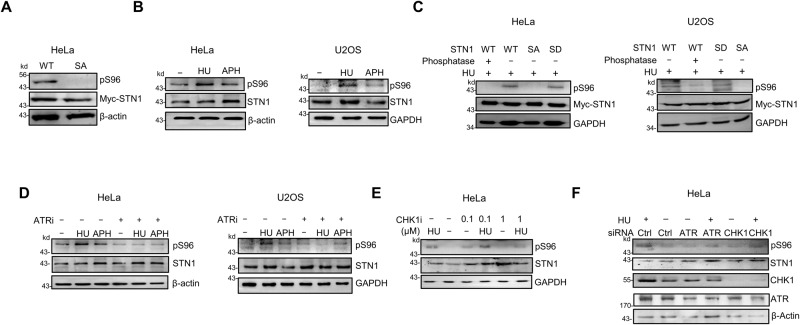

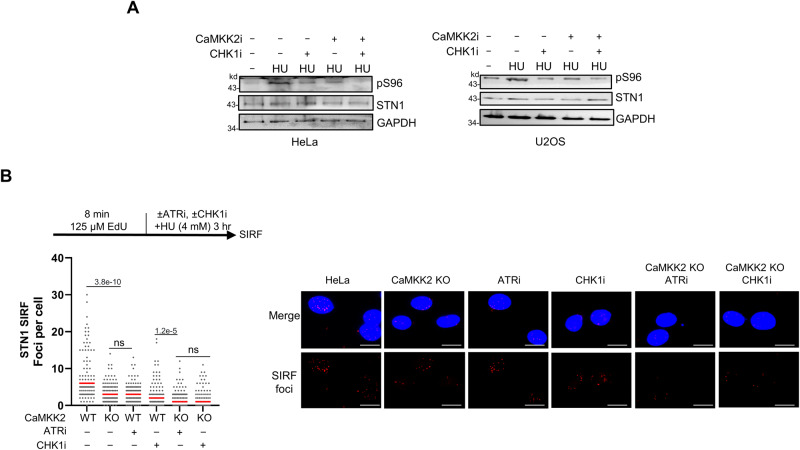

References
-
- Macheret, M. & Halazonetis, T. D. DNA Replication Stress as a Hallmark of Cancer. Annual Review of Pathology: Mechanisms of Disease10, 425–448 (2015). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
