

Mapping the dynamic genetic regulatory architecture of HLA genes at single-cell resolution
- PMID: 38036787
- PMCID: PMC10787945
- DOI: 10.1038/s41588-023-01586-6
Mapping the dynamic genetic regulatory architecture of HLA genes at single-cell resolution
Abstract
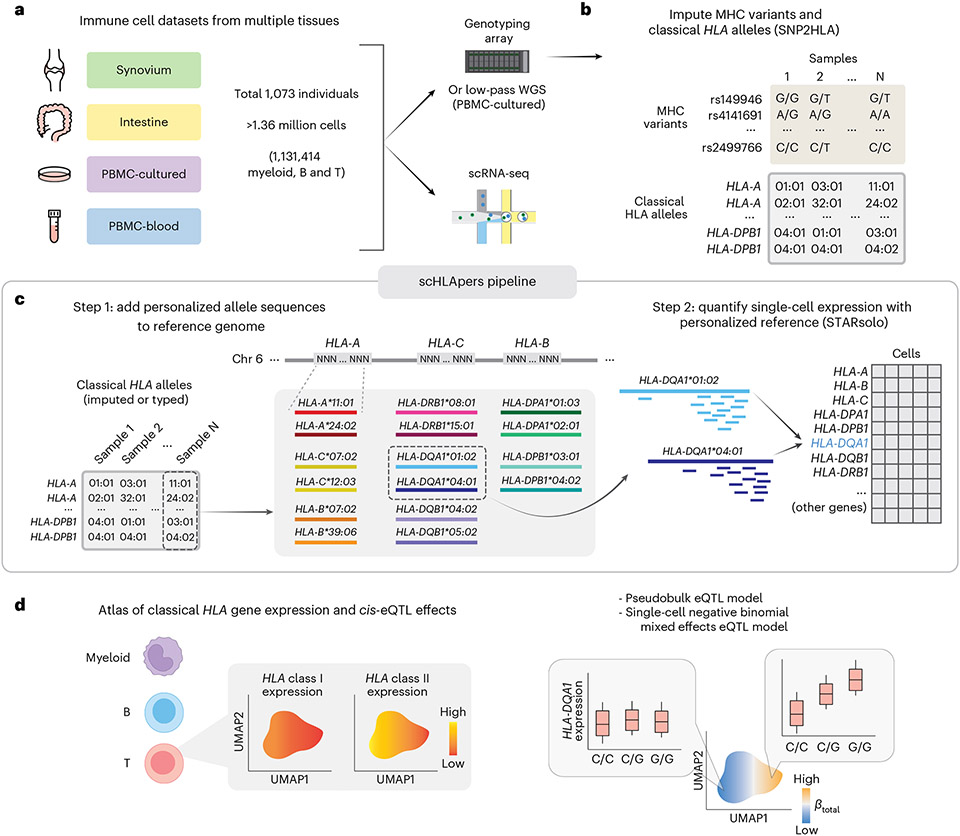
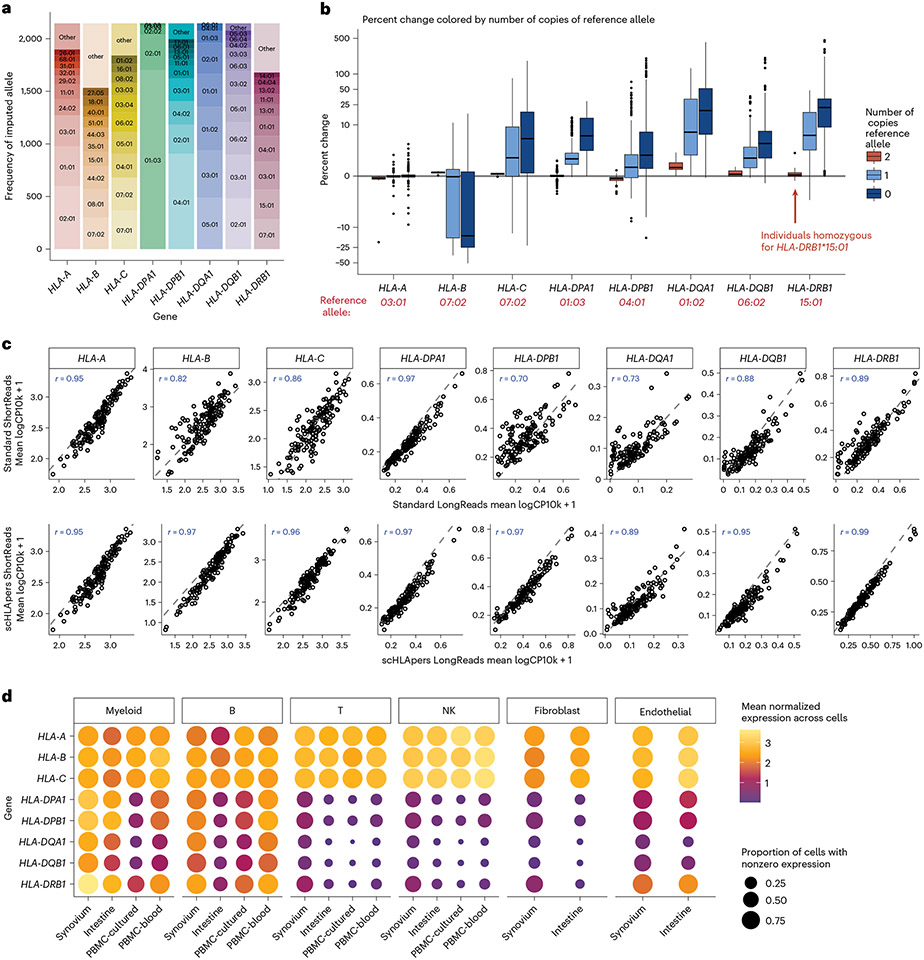
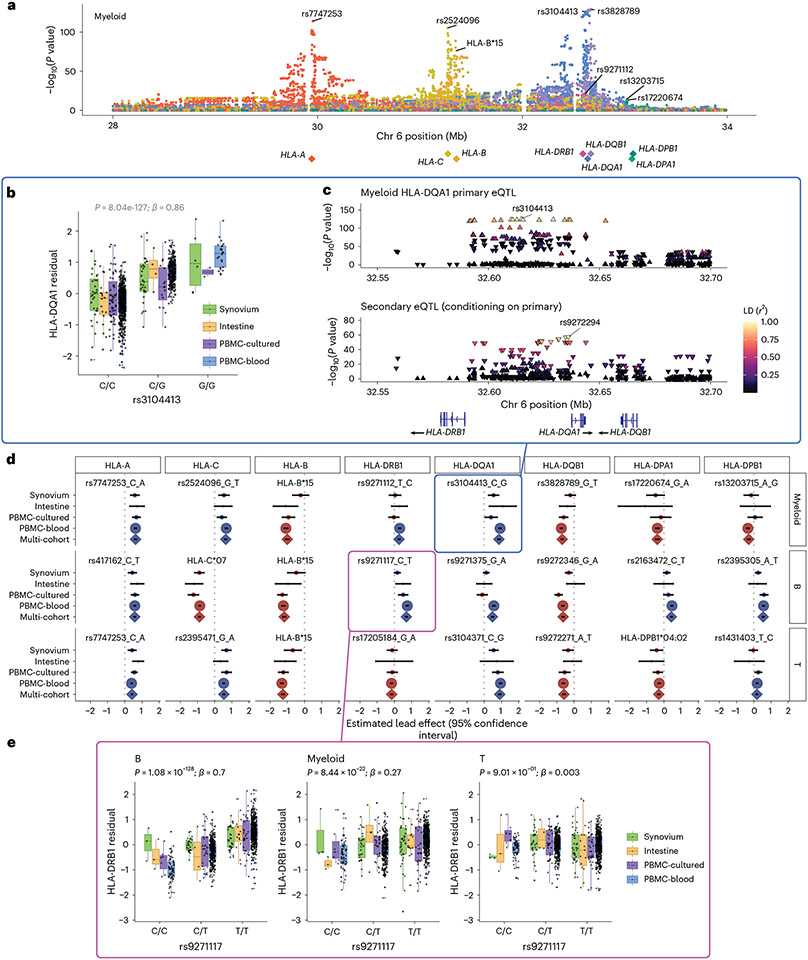
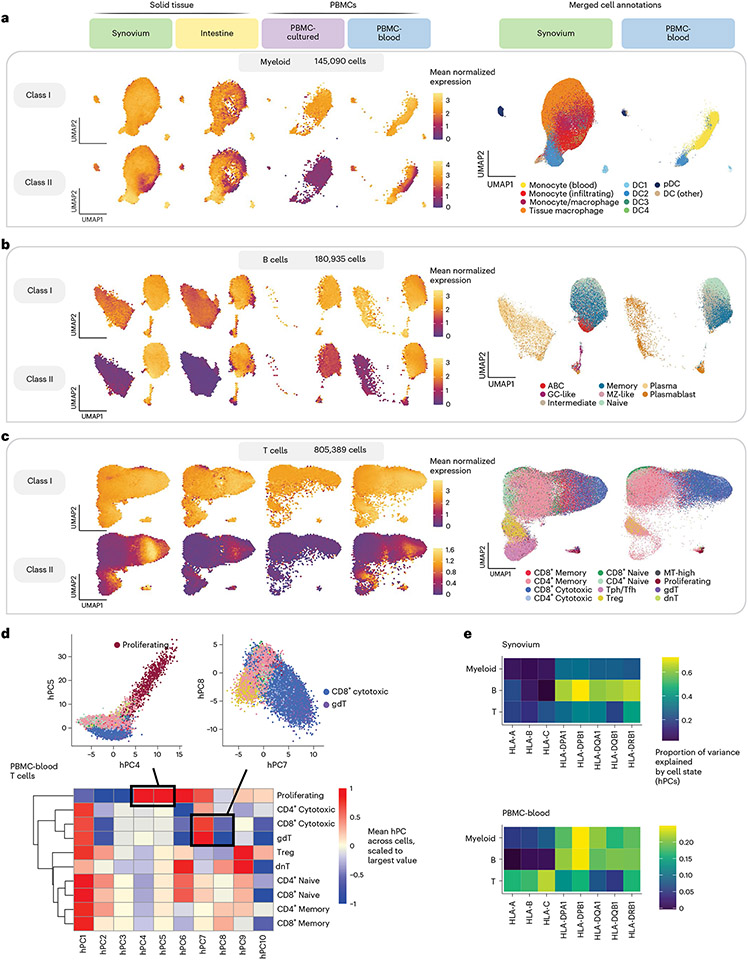
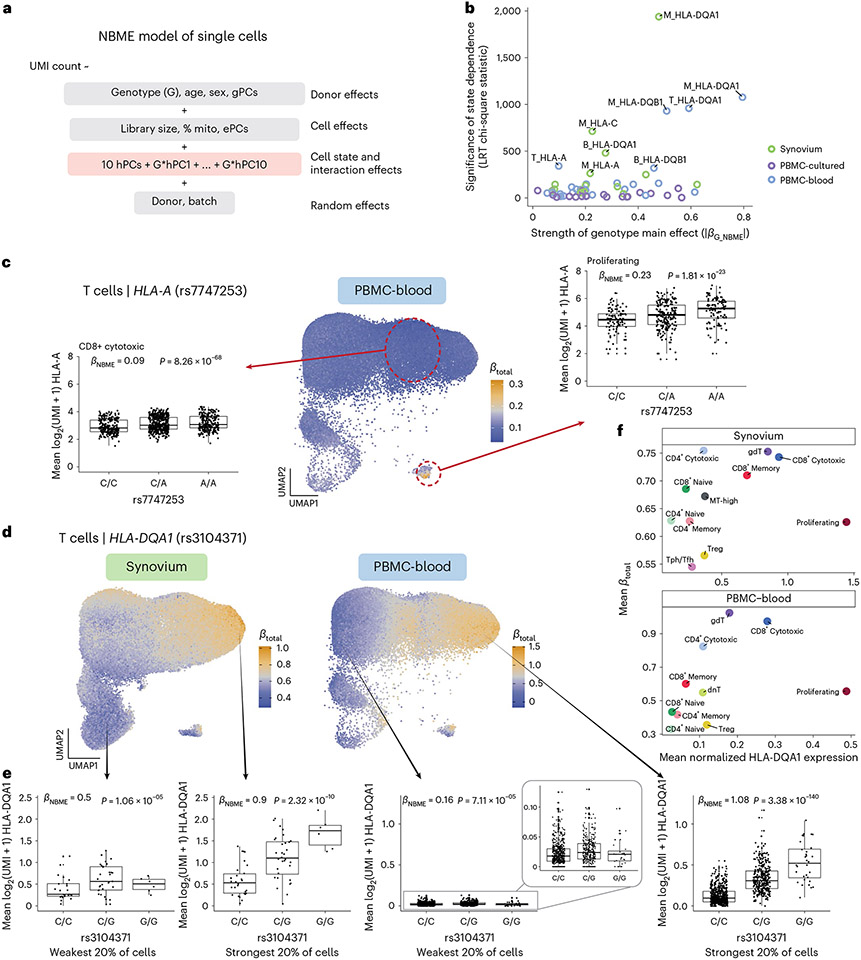
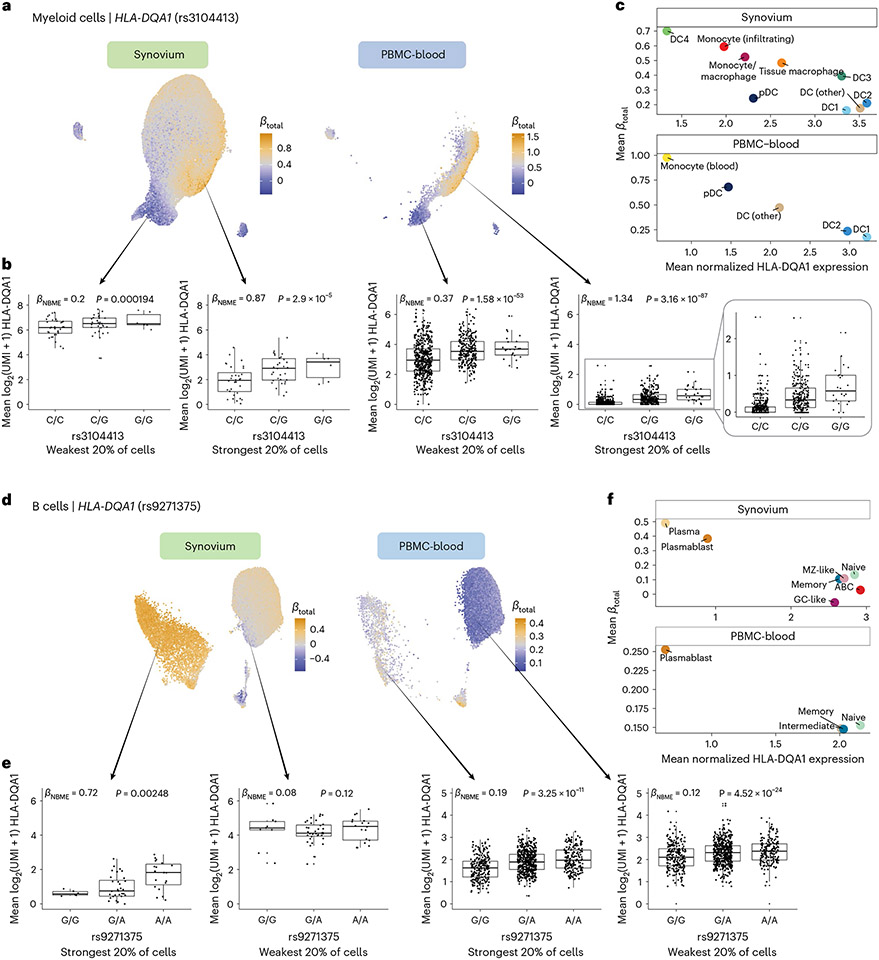
The human leukocyte antigen (HLA) locus plays a critical role in complex traits spanning autoimmune and infectious diseases, transplantation and cancer. While coding variation in HLA genes has been extensively documented, regulatory genetic variation modulating HLA expression levels has not been comprehensively investigated. Here we mapped expression quantitative trait loci (eQTLs) for classical HLA genes across 1,073 individuals and 1,131,414 single cells from three tissues. To mitigate technical confounding, we developed scHLApers, a pipeline to accurately quantify single-cell HLA expression using personalized reference genomes. We identified cell-type-specific cis-eQTLs for every classical HLA gene. Modeling eQTLs at single-cell resolution revealed that many eQTL effects are dynamic across cell states even within a cell type. HLA-DQ genes exhibit particularly cell-state-dependent effects within myeloid, B and T cells. For example, a T cell HLA-DQA1 eQTL ( rs3104371 ) is strongest in cytotoxic cells. Dynamic HLA regulation may underlie important interindividual variability in immune responses.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
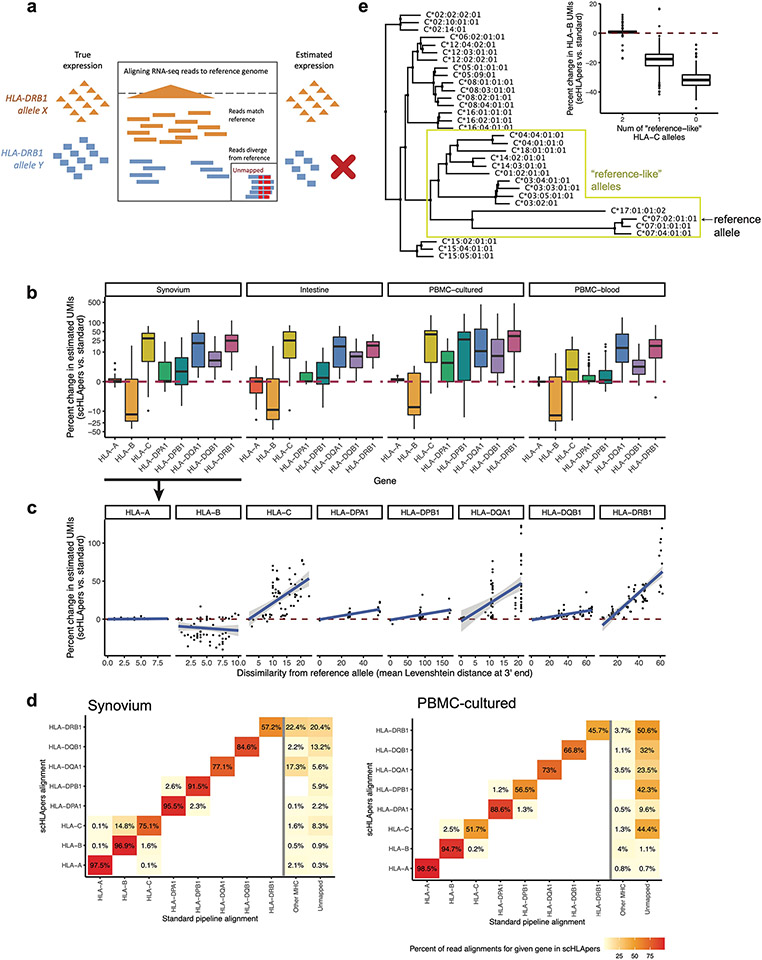
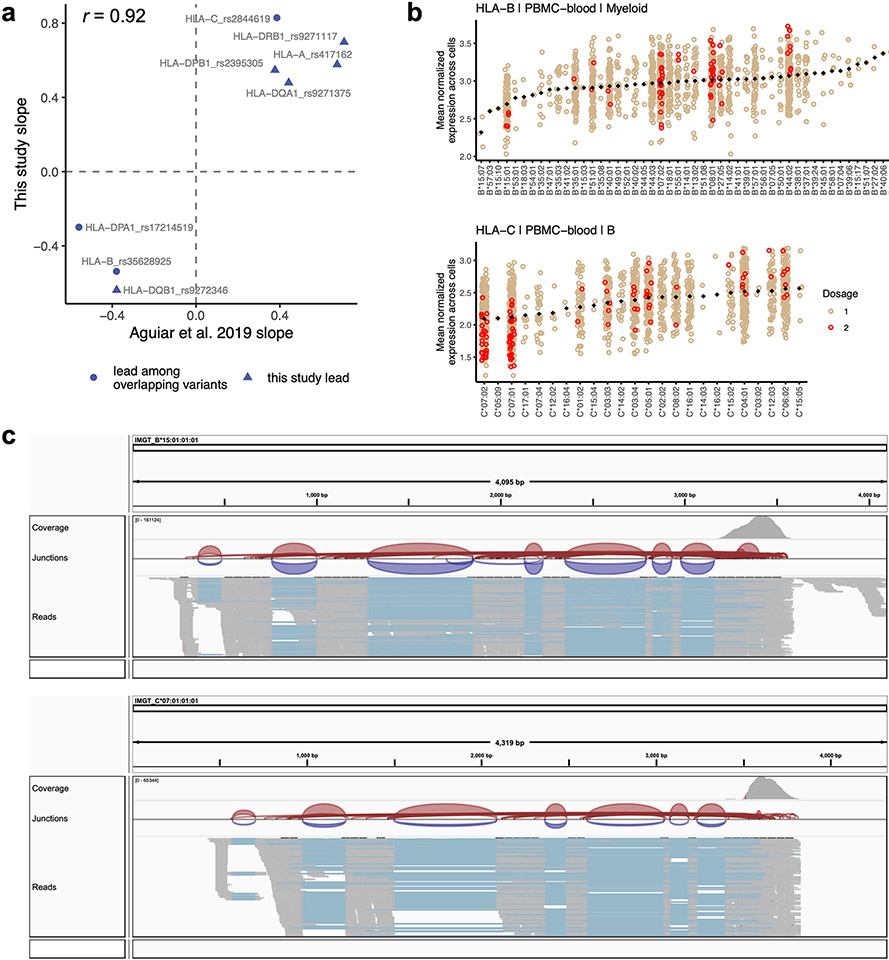
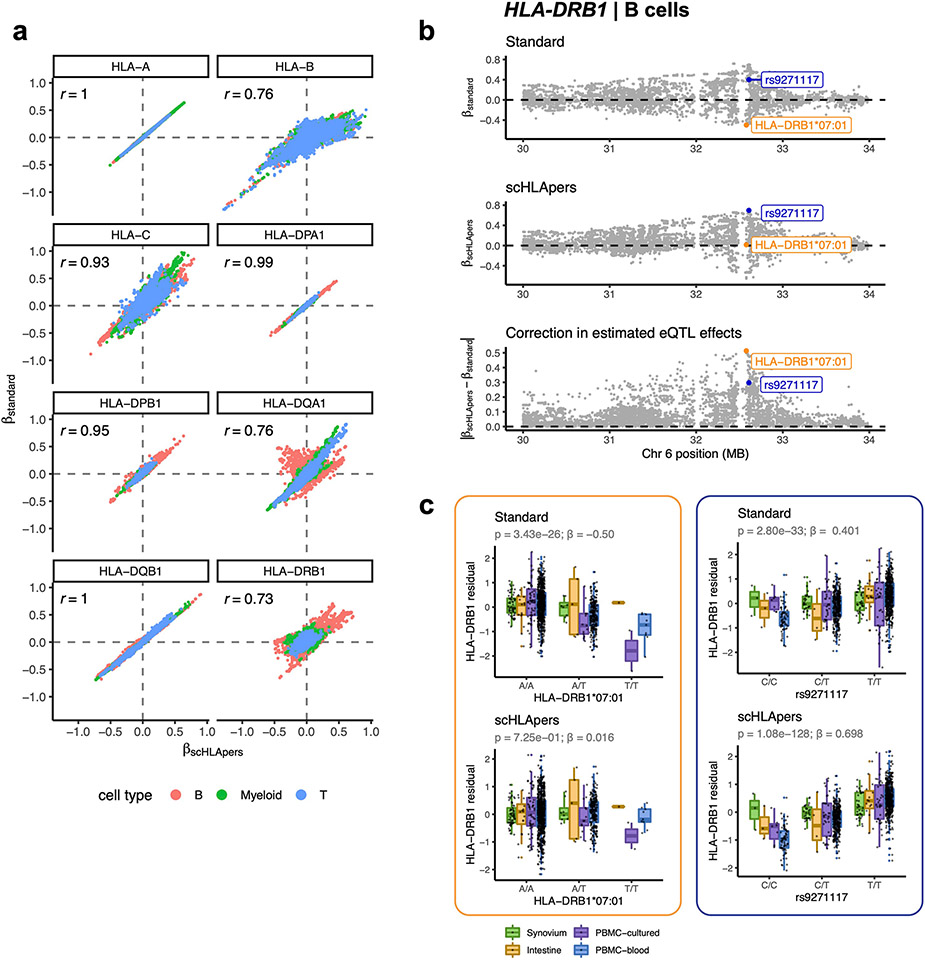
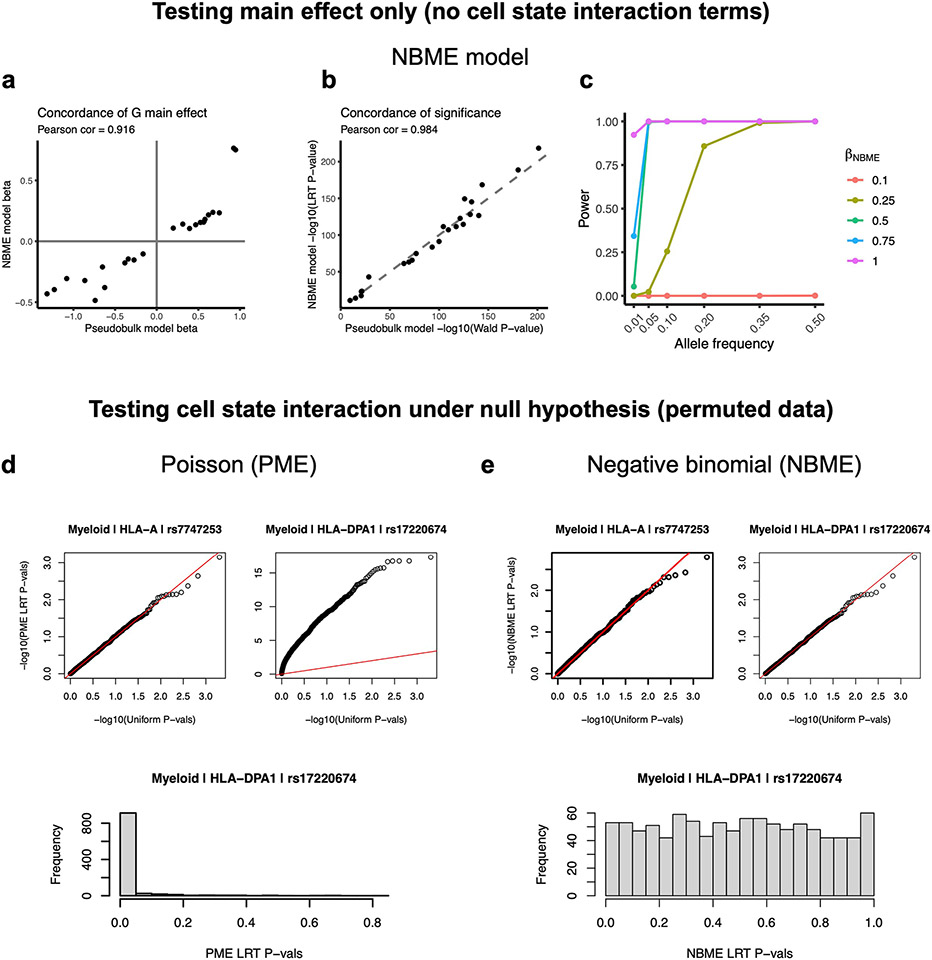
Update of
-
Mapping the dynamic genetic regulatory architecture of HLA genes at single-cell resolution.medRxiv [Preprint]. 2023 Mar 20:2023.03.14.23287257. doi: 10.1101/2023.03.14.23287257. medRxiv. 2023. Update in: Nat Genet. 2023 Dec;55(12):2255-2268. doi: 10.1038/s41588-023-01586-6. PMID: 36993194 Free PMC article. Updated. Preprint.
References
-
- Dendrou CA, Petersen J, Rossjohn J & Fugger L HLA variation and disease. Nat. Rev. Immunol 18, 325–339 (2018). - PubMed
Publication types
MeSH terms
Grants and funding
- P01 AI148102/AI/NIAID NIH HHS/United States
- P30 DK043351/DK/NIDDK NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- T32 GM144273/GM/NIGMS NIH HHS/United States
- R01 AR063759/AR/NIAMS NIH HHS/United States
- UH2 AR067677/AR/NIAMS NIH HHS/United States
- K08 AR077037/AR/NIAMS NIH HHS/United States
- U01 HG012009/HG/NHGRI NIH HHS/United States
- F30 AI172238/AI/NIAID NIH HHS/United States
- F30 AI157385/AI/NIAID NIH HHS/United States
- MR/M007669/1/MRC_/Medical Research Council/United Kingdom
- R56 HG013083/HG/NHGRI NIH HHS/United States
- P30 AR070253/AR/NIAMS NIH HHS/United States
- UC2 AR081023/AR/NIAMS NIH HHS/United States
- T32 HG002295/HG/NHGRI NIH HHS/United States
- T32 GM007753/GM/NIGMS NIH HHS/United States
- T32 AR007530/AR/NIAMS NIH HHS/United States
- MR/P021220/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Research Materials
