

Neurological disease in xeroderma pigmentosum: prospective cohort study of its features and progression
- PMID: 38040034
- PMCID: PMC10690019
- DOI: 10.1093/brain/awad266
Neurological disease in xeroderma pigmentosum: prospective cohort study of its features and progression
Abstract
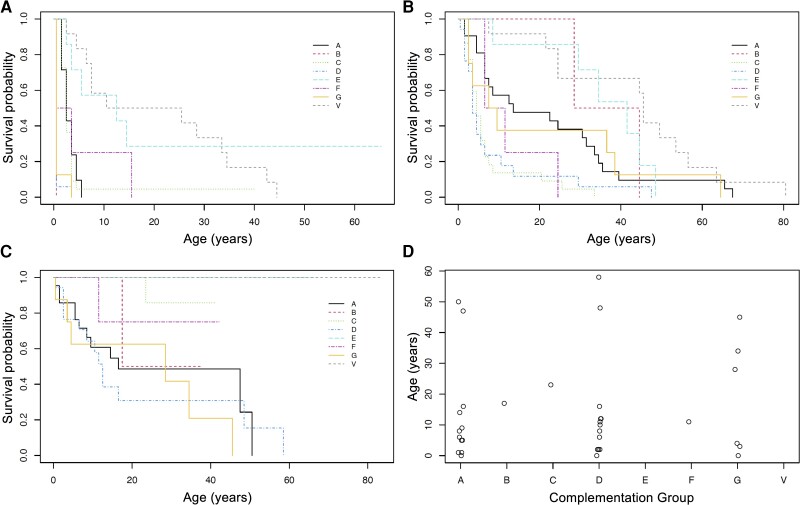
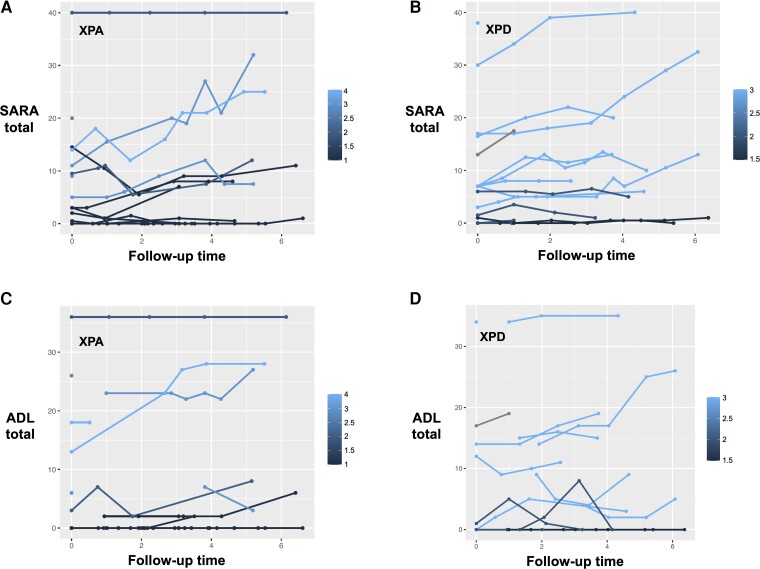
Xeroderma pigmentosum (XP) results from biallelic mutations in any of eight genes involved in DNA repair systems, thus defining eight different genotypes (XPA, XPB, XPC, XPD, XPE, XPF, XPG and XP variant or XPV). In addition to cutaneous and ophthalmological features, some patients present with XP neurological disease. It is unknown whether the different neurological signs and their progression differ among groups. Therefore, we aim to characterize the XP neurological disease and its evolution in the heterogeneous UK XP cohort. Patients with XP were followed in the UK National XP Service, from 2009 to 2021. Age of onset for different events was recorded. Cerebellar ataxia and additional neurological signs and symptoms were rated with the Scale for the Assessment and Rating of Ataxia (SARA), the Inventory of Non-Ataxia Signs (INAS) and the Activities of Daily Living questionnaire (ADL). Patients' mutations received scores based on their predicted effects. Data from available ancillary tests were collected. Ninety-three XP patients were recruited. Thirty-six (38.7%) reported neurological symptoms, especially in the XPA, XPD and XPG groups, with early-onset and late-onset forms, and typically appearing after cutaneous and ophthalmological symptoms. XPA, XPD and XPG patients showed higher SARA scores compared to XPC, XPE and XPV. SARA total scores significantly increased over time in XPD (0.91 points/year, 95% confidence interval: 0.61, 1.21) and XPA (0.63 points/year, 95% confidence interval: 0.38, 0.89). Hyporeflexia, hypopallesthaesia, upper motor neuron signs, chorea, dystonia, oculomotor signs and cognitive impairment were frequent findings in XPA, XPD and XPG. Cerebellar and global brain atrophy, axonal sensory and sensorimotor neuropathies, and sensorineural hearing loss were common findings in patients. Some XPC, XPE and XPV cases presented with abnormalities on examination and/or ancillary tests, suggesting underlying neurological involvement. More severe mutations were associated with a faster progression in SARA total score in XPA (0.40 points/year per 1-unit increase in severity score) and XPD (0.60 points/year per 1-unit increase), and in ADL total score in XPA (0.35 points/year per 1-unit increase). Symptomatic and asymptomatic forms of neurological disease are frequent in XP patients, and neurological symptoms can be an important cause of disability. Typically, the neurological disease will be preceded by cutaneous and ophthalmological features, and these should be actively searched in patients with idiopathic late-onset neurological syndromes. Scales assessing cerebellar function, especially walking and speech, and disability can show progression in some of the groups. Mutation severity can be used as a prognostic biomarker for stratification purposes in clinical trials.
Keywords: DNA repair; Huntington’s disease-like phenotypes; biomarkers; cerebellar ataxia; neurocutaneous syndromes; nucleotide excision repair.
© The Author(s) 2023. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
P.G. has received grants and honoraria for advisory board from Vico Therapeutics, honoraria for advisory board from Triplet Therapeutics, grants and personal fees from Reata Pharmaceutical, grants from Wave. The other authors report no other competing interests.
Figures
References
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
