

Haplotype-based inference of recent effective population size in modern and ancient DNA samples
- PMID: 38040695
- PMCID: PMC10692198
- DOI: 10.1038/s41467-023-43522-6
Haplotype-based inference of recent effective population size in modern and ancient DNA samples
Abstract
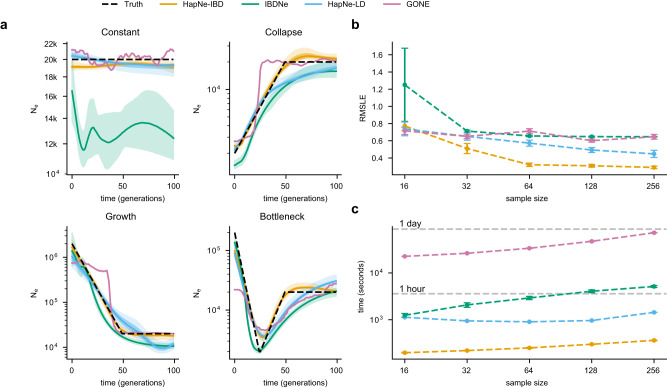
Individuals sharing recent ancestors are likely to co-inherit large identical-by-descent (IBD) genomic regions. The distribution of these IBD segments in a population may be used to reconstruct past demographic events such as effective population size variation, but accurate IBD detection is difficult in ancient DNA data and in underrepresented populations with limited reference data. In this work, we introduce an accurate method for inferring effective population size variation during the past ~2000 years in both modern and ancient DNA data, called HapNe. HapNe infers recent population size fluctuations using either IBD sharing (HapNe-IBD) or linkage disequilibrium (HapNe-LD), which does not require phasing and can be computed in low coverage data, including data sets with heterogeneous sampling times. HapNe shows improved accuracy in a range of simulated demographic scenarios compared to currently available methods for IBD-based and LD-based inference of recent effective population size, while requiring fewer computational resources. We apply HapNe to several modern populations from the 1,000 Genomes Project, the UK Biobank, the Allen Ancient DNA Resource, and recently published samples from Iron Age Britain, detecting multiple instances of recent effective population size variation across these groups.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- Charlesworth, B. Effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet.10, 195–205 (2009). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
