

Accurate de novo peptide sequencing using fully convolutional neural networks
- PMID: 38042873
- PMCID: PMC10693636
- DOI: 10.1038/s41467-023-43010-x
Accurate de novo peptide sequencing using fully convolutional neural networks
Abstract
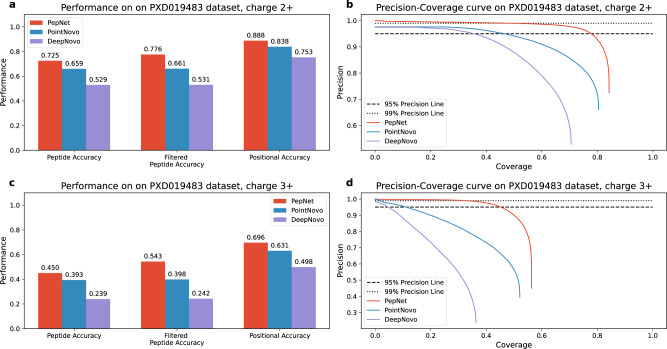
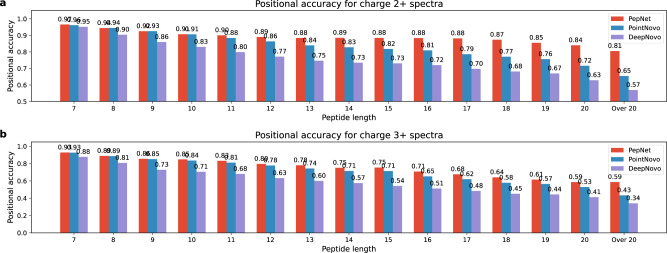
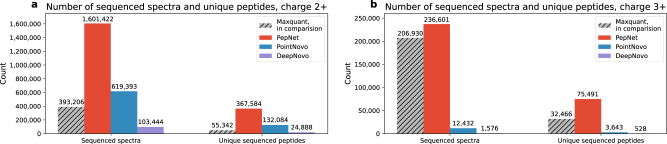
De novo peptide sequencing, which does not rely on a comprehensive target sequence database, provides us with a way to identify novel peptides from tandem mass spectra. However, current de novo sequencing algorithms suffer from low accuracy and coverage, which hinders their application in proteomics. In this paper, we present PepNet, a fully convolutional neural network for high accuracy de novo peptide sequencing. PepNet takes an MS/MS spectrum (represented as a high-dimensional vector) as input, and outputs the optimal peptide sequence along with its confidence score. The PepNet model is trained using a total of 3 million high-energy collisional dissociation MS/MS spectra from multiple human peptide spectral libraries. Evaluation results show that PepNet significantly outperforms current best-performing de novo sequencing algorithms (e.g. PointNovo and DeepNovo) in both peptide-level accuracy and positional-level accuracy. PepNet can sequence a large fraction of spectra that were not identified by database search engines, and thus could be used as a complementary tool to database search engines for peptide identification in proteomics. In addition, PepNet runs around 3x and 7x faster than PointNovo and DeepNovo on GPUs, respectively, thus being more suitable for the analysis of large-scale proteomics data.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures







References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
