

Non-canonical STING-PERK pathway dependent epigenetic regulation of vascular endothelial dysfunction via integrating IRF3 and NF- κ B in inflammatory response
- PMID: 38045042
- PMCID: PMC10692388
- DOI: 10.1016/j.apsb.2023.08.015
Non-canonical STING-PERK pathway dependent epigenetic regulation of vascular endothelial dysfunction via integrating IRF3 and NF- κ B in inflammatory response
Abstract
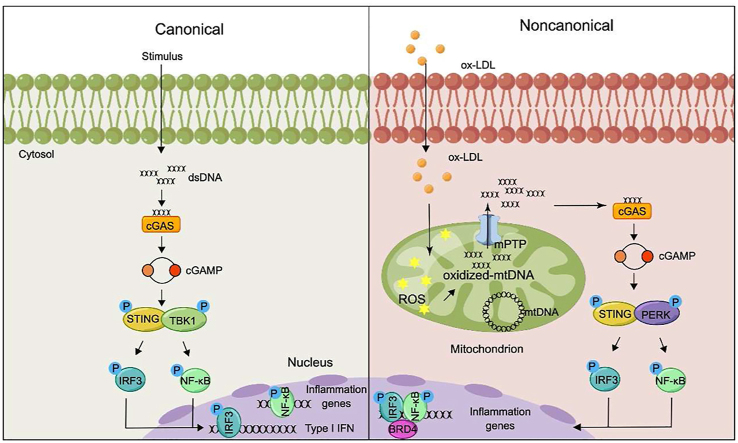
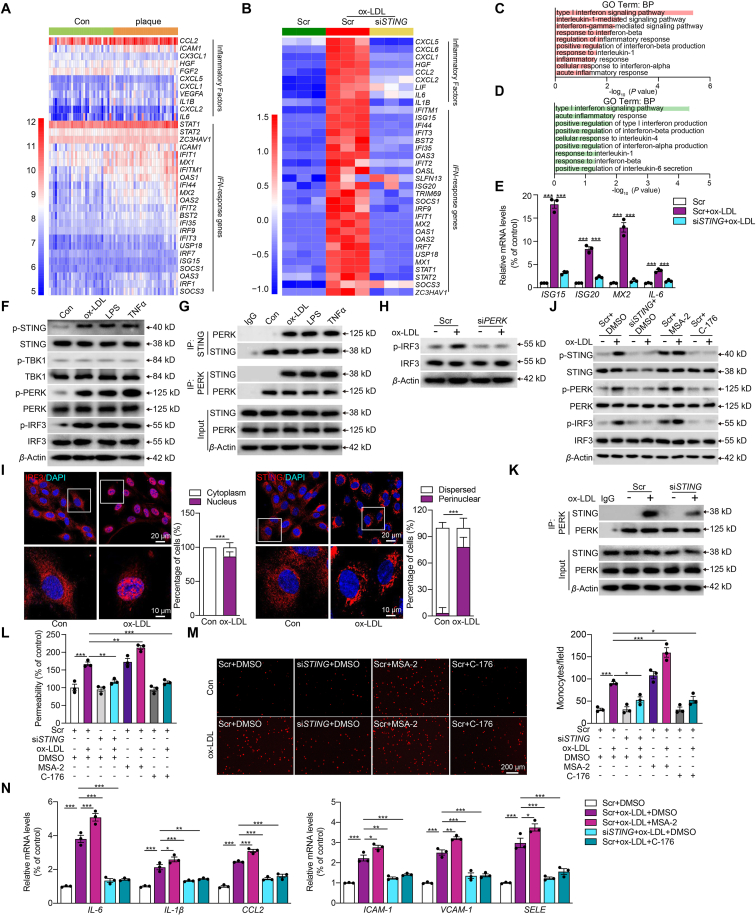
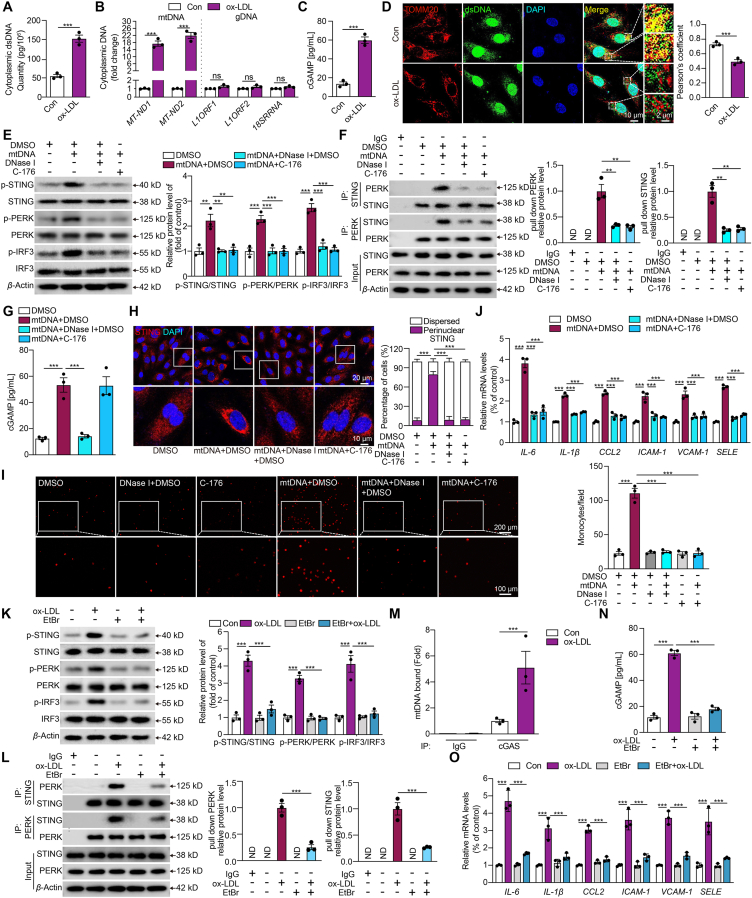
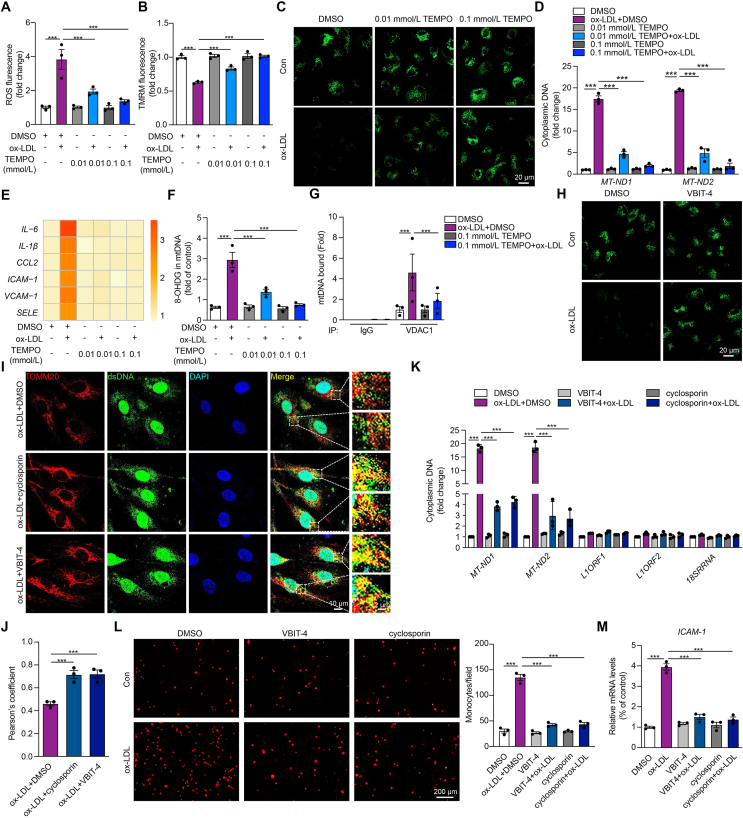
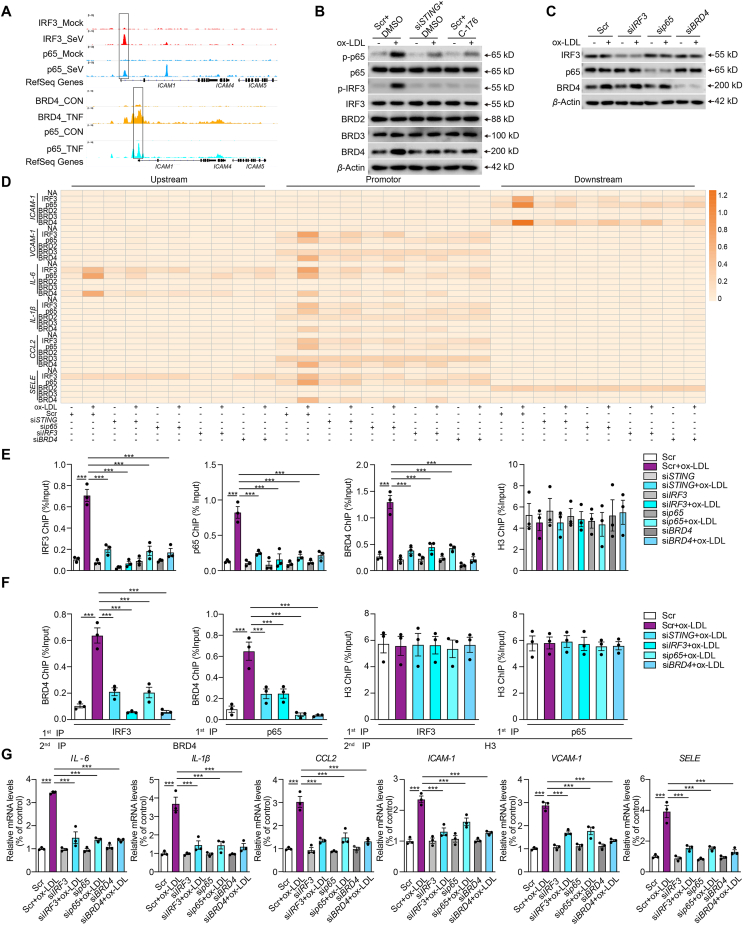
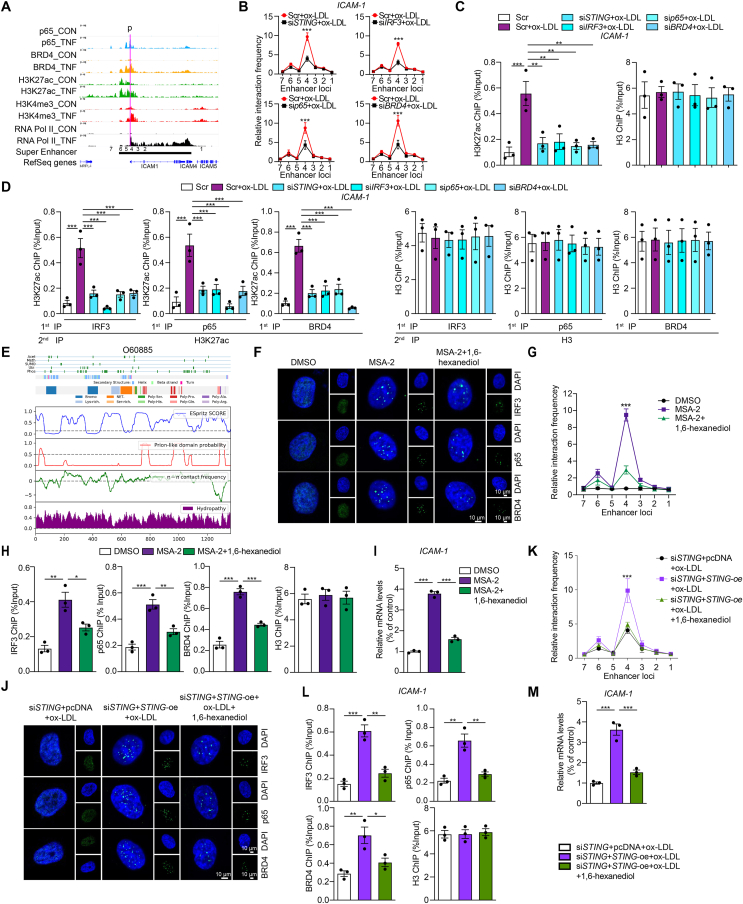
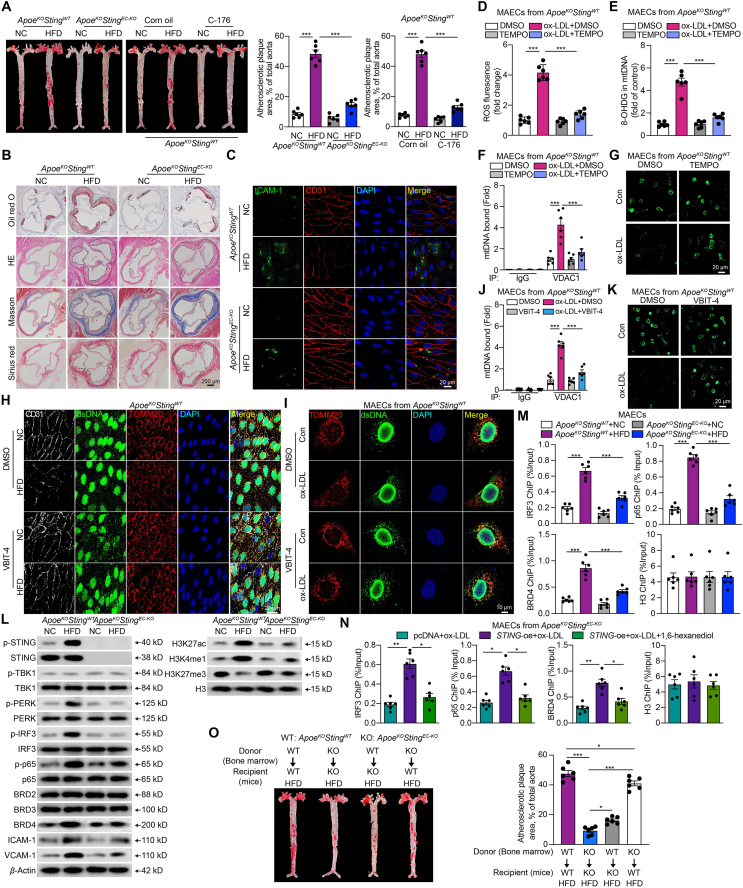
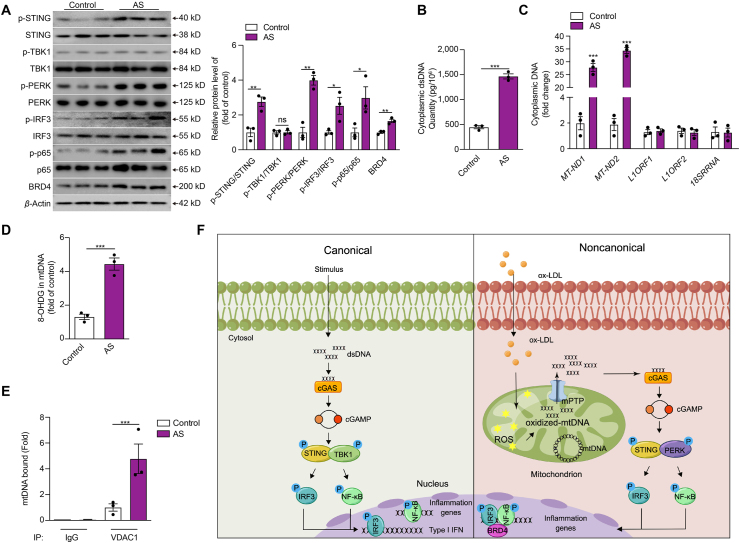
Inflammation-driven endothelial dysfunction is the major initiating factor in atherosclerosis, while the underlying mechanism remains elusive. Here, we report that the non-canonical stimulator of interferon genes (STING)-PKR-like ER kinase (PERK) pathway was significantly activated in both human and mice atherosclerotic arteries. Typically, STING activation leads to the activation of interferon regulatory factor 3 (IRF3) and nuclear factor-kappa B (NF-κB)/p65, thereby facilitating IFN signals and inflammation. In contrast, our study reveals the activated non-canonical STING-PERK pathway increases scaffold protein bromodomain protein 4 (BRD4) expression, which encourages the formation of super-enhancers on the proximal promoter regions of the proinflammatory cytokines, thereby enabling the transactivation of these cytokines by integrating activated IRF3 and NF-κB via a condensation process. Endothelium-specific STING and BRD4 deficiency significantly decreased the plaque area and inflammation. Mechanistically, this pathway is triggered by leaked mitochondrial DNA (mtDNA) via mitochondrial permeability transition pore (mPTP), formed by voltage-dependent anion channel 1 (VDAC1) oligomer interaction with oxidized mtDNA upon cholesterol oxidation stimulation. Especially, compared to macrophages, endothelial STING activation plays a more pronounced role in atherosclerosis. We propose a non-canonical STING-PERK pathway-dependent epigenetic paradigm in atherosclerosis that integrates IRF3, NF-κB and BRD4 in inflammatory responses, which provides emerging therapeutic modalities for vascular endothelial dysfunction.
Keywords: Atherosclerosis; BRD4; Endothelial dysfunction; Inflammation; Mitochondrial DNA; PERK; ROS; STING.
© 2023 Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences. Production and hosting by Elsevier B.V.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Benjamin E.J., Virani S.S., Callaway C.W., Chamberlain A.M., Chang A.R., Cheng S., et al. Heart disease and stroke statistics-2018 update: a report from the American Heart Association. Circulation. 2018;137:e67–e492. - PubMed
-
- Xu S., Ilyas I., Little P.J., Li H., Kamato D., Zheng X., et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: from mechanism to pharmacotherapies. Pharmacol Rev. 2021;73:924–967. - PubMed
-
- Zhang X., Bai X.C., Chen Z.J. Structures and mechanisms in the cGAS–STING innate immunity pathway. Immunity. 2020;53:43–53. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
