

MYCN and SNRPD3 cooperate to maintain a balance of alternative splicing events that drives neuroblastoma progression
- PMID: 38049564
- PMCID: PMC10824661
- DOI: 10.1038/s41388-023-02897-y
MYCN and SNRPD3 cooperate to maintain a balance of alternative splicing events that drives neuroblastoma progression
Abstract
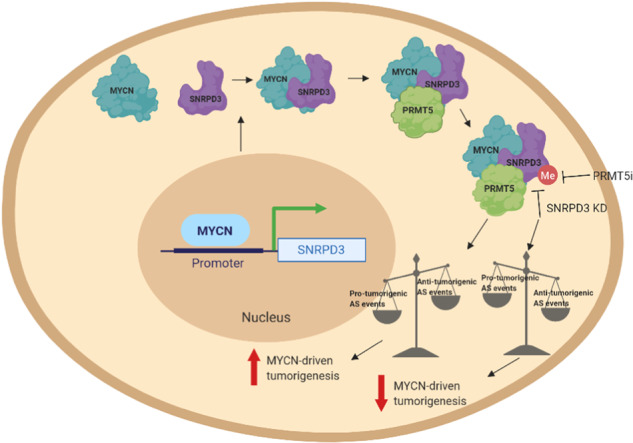
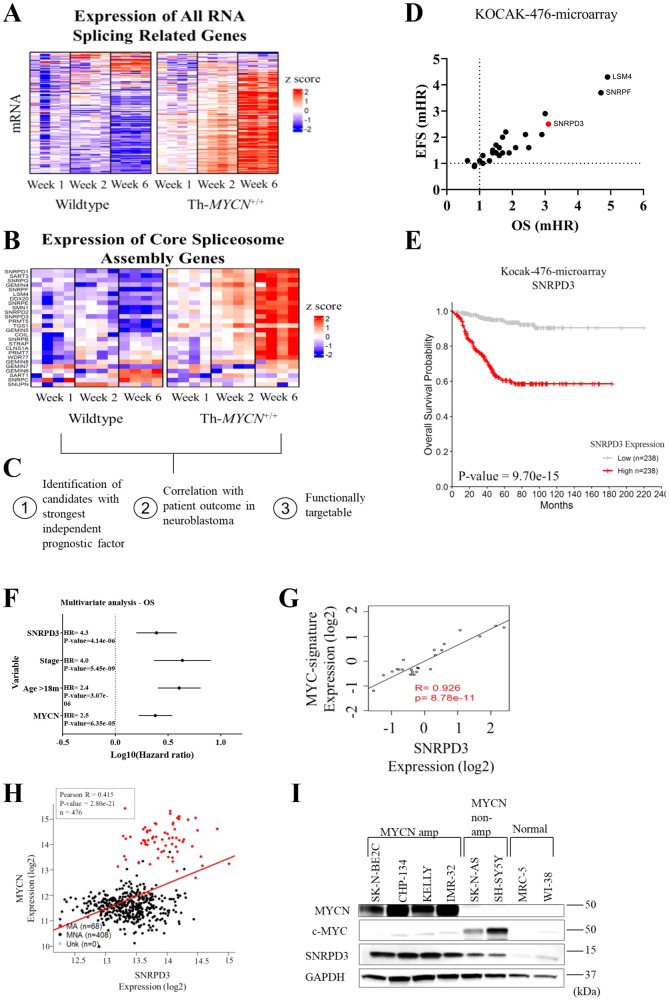
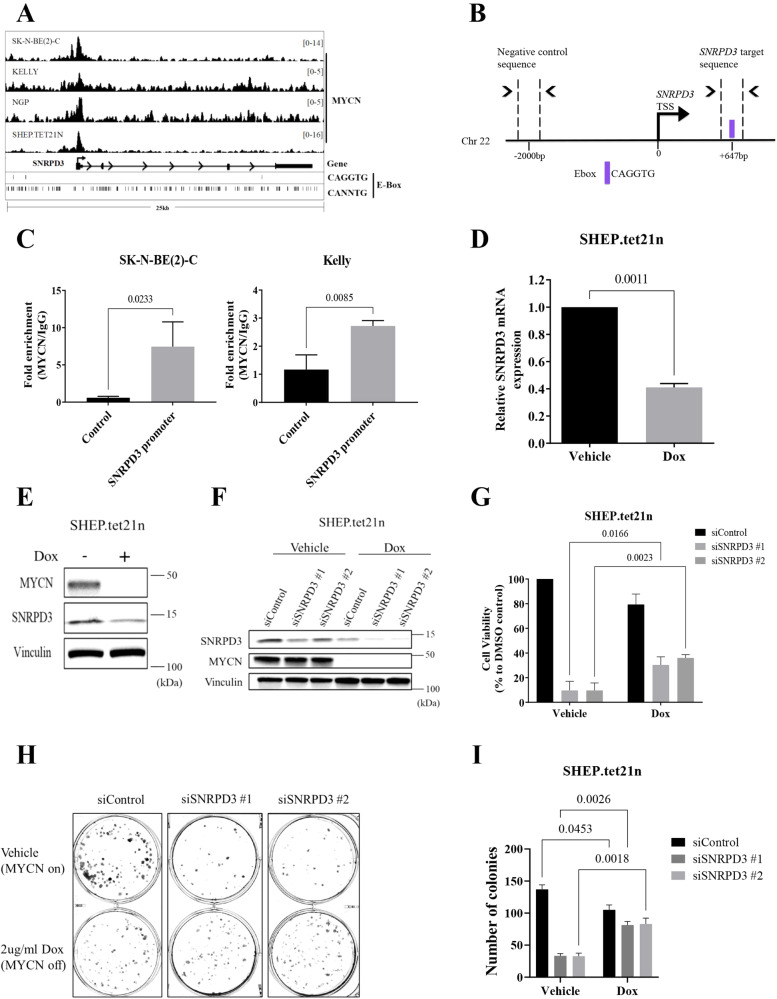
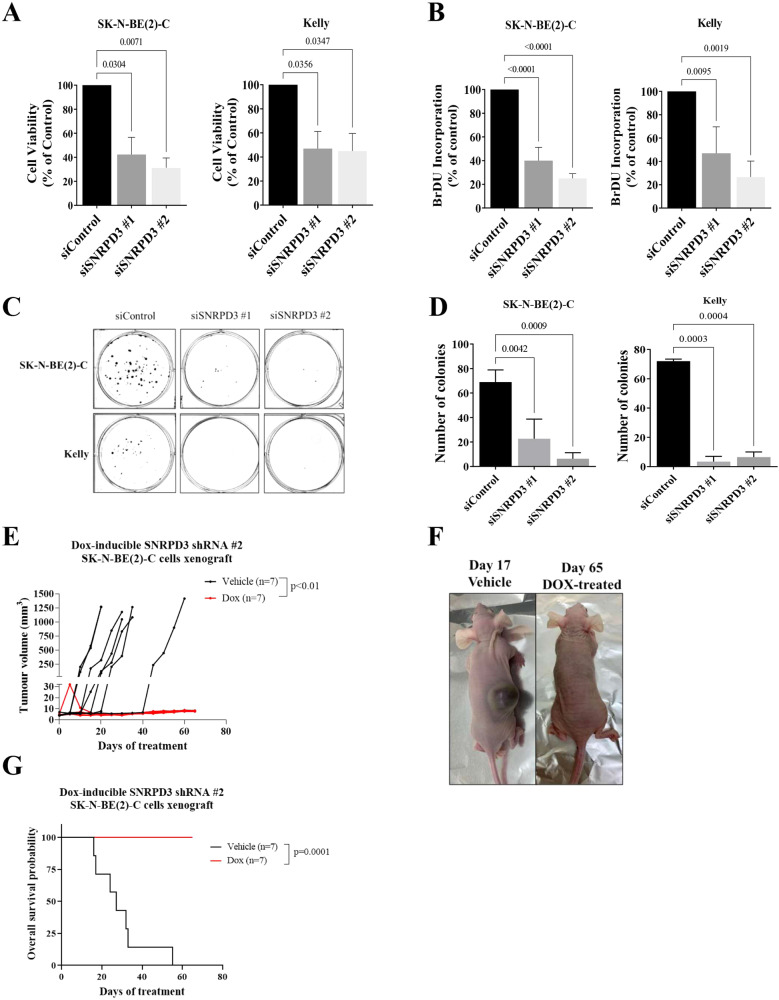
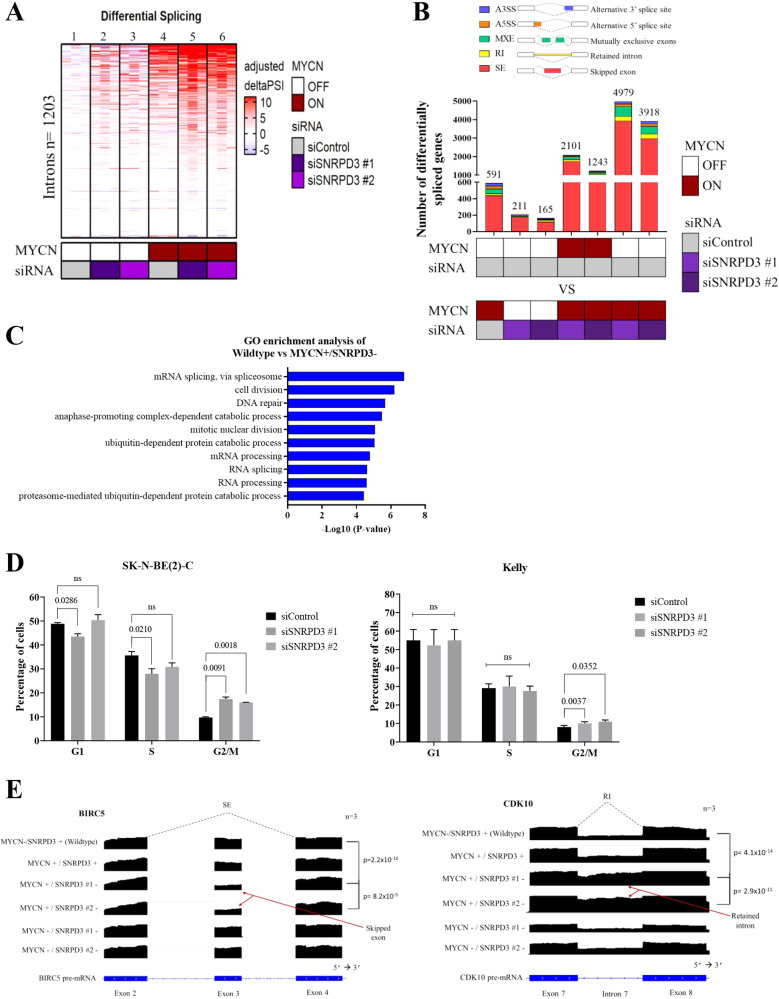
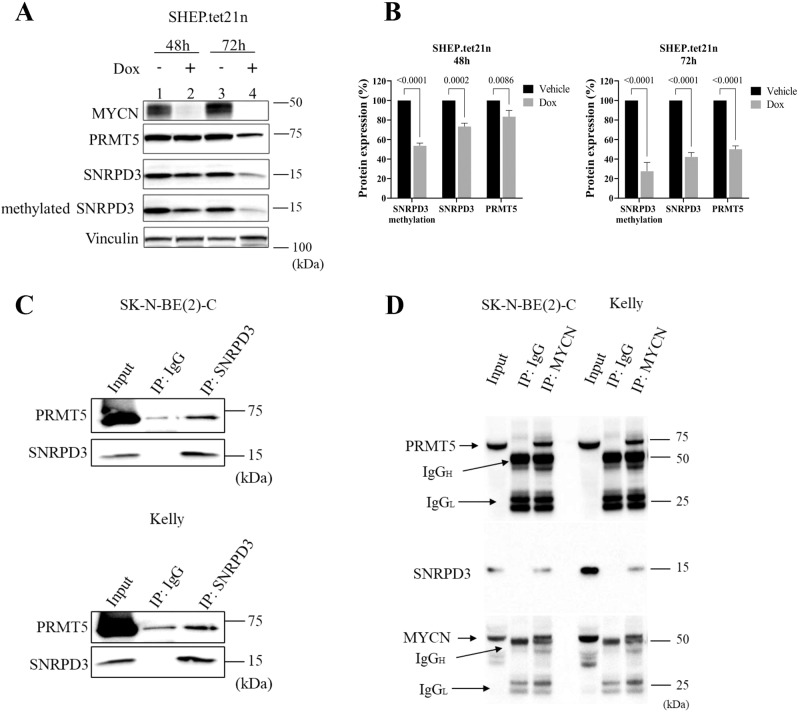
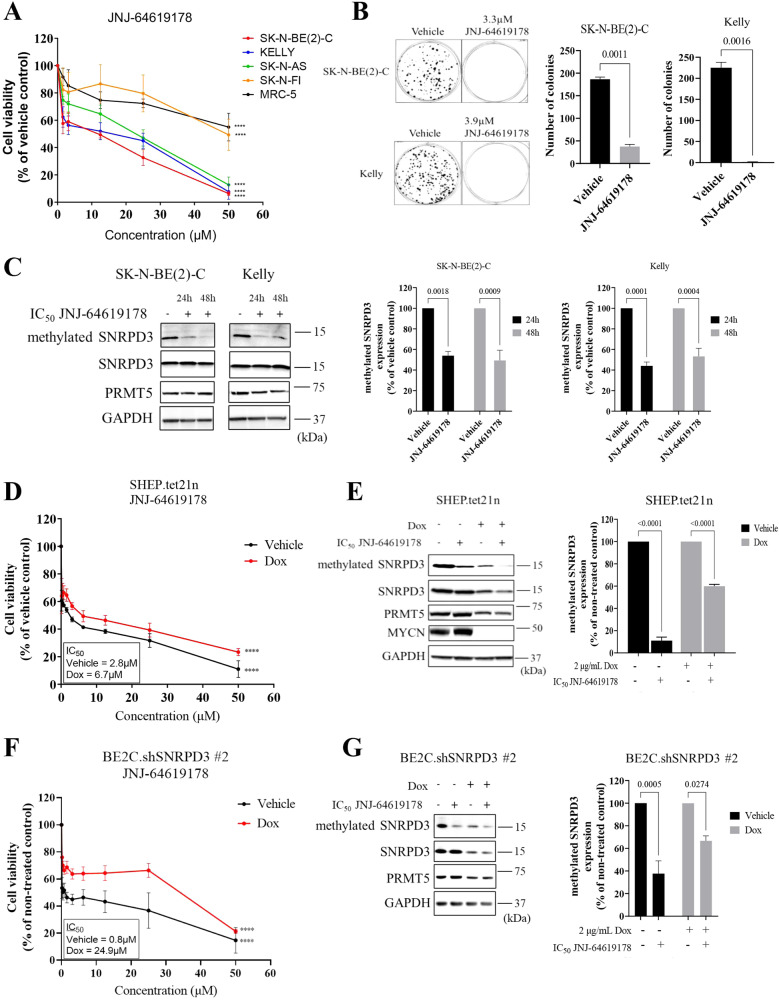
Many of the pro-tumorigenic functions of the oncogene MYCN are attributed to its regulation of global gene expression programs. Alternative splicing is another important regulator of gene expression and has been implicated in neuroblastoma development, however, the molecular mechanisms remain unknown. We found that MYCN up-regulated the expression of the core spliceosomal protein, SNRPD3, in models of neuroblastoma initiation and progression. High mRNA expression of SNRPD3 in human neuroblastoma tissues was a strong, independent prognostic factor for poor patient outcome. Repression of SNRPD3 expression correlated with loss of colony formation in vitro and reduced tumorigenicity in vivo. The effect of SNRPD3 on cell viability was in part dependent on MYCN as an oncogenic co-factor. RNA-sequencing revealed a global increase in the number of genes being differentially spliced when MYCN was overexpressed. Surprisingly, depletion of SNRPD3 in the presence of overexpressed MYCN further increased differential splicing, particularly of cell cycle regulators, such as BIRC5 and CDK10. MYCN directly bound SNRPD3, and the protein arginine methyltransferase, PRMT5, consequently increasing SNRPD3 methylation. Indeed, the PRMT5 inhibitor, JNJ-64619178, reduced cell viability and SNRPD3 methylation in neuroblastoma cells with high SNRPD3 and MYCN expression. Our findings demonstrate a functional relationship between MYCN and SNRPD3, which maintains the fidelity of MYCN-driven alternative splicing in the narrow range required for neuroblastoma cell growth. SNRPD3 methylation and its protein-protein interface with MYCN represent novel therapeutic targets. Hypothetical model for SNRPD3 as a co-factor for MYCN oncogenesis. SNRPD3 and MYCN participate in a regulatory loop to balance splicing fidelity in neuroblastoma cells. First MYCN transactivates SNRPD3 to lead to high-level expression. Second, SNRPD3 and MYCN form a protein complex involving PRMT5. Third, this leads to balanced alterative splicing (AS) activitiy that is favorable to neuroblastoma. Together this forms as a therapeutic vulnerability where SNRPD3 perturbation or PRMT5 inhibitors are selectively toxic to neuroblastoma by conditionally disturbing splicing activity.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
