

Perturbational phenotyping of human blood cells reveals genetically determined latent traits associated with subsets of common diseases
- PMID: 38049662
- PMCID: PMC10786715
- DOI: 10.1038/s41588-023-01600-x
Perturbational phenotyping of human blood cells reveals genetically determined latent traits associated with subsets of common diseases
Abstract
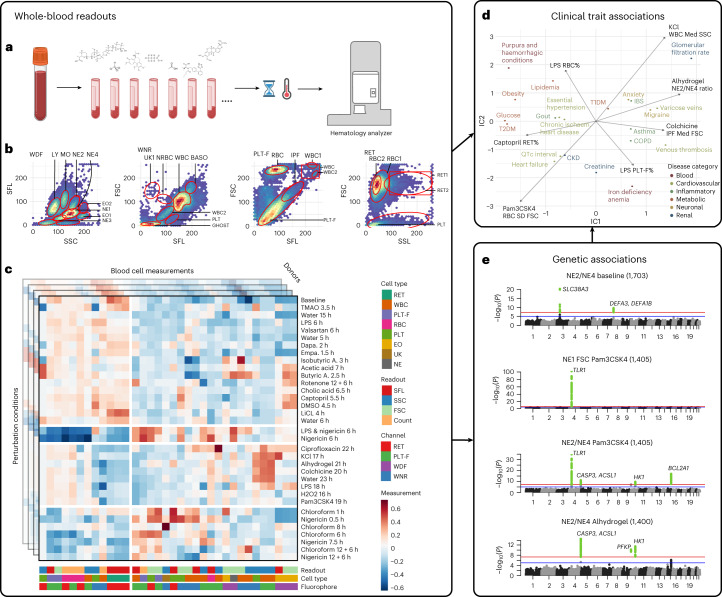
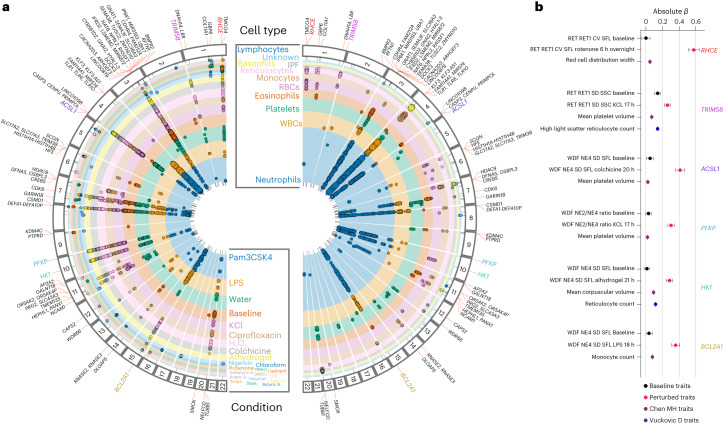
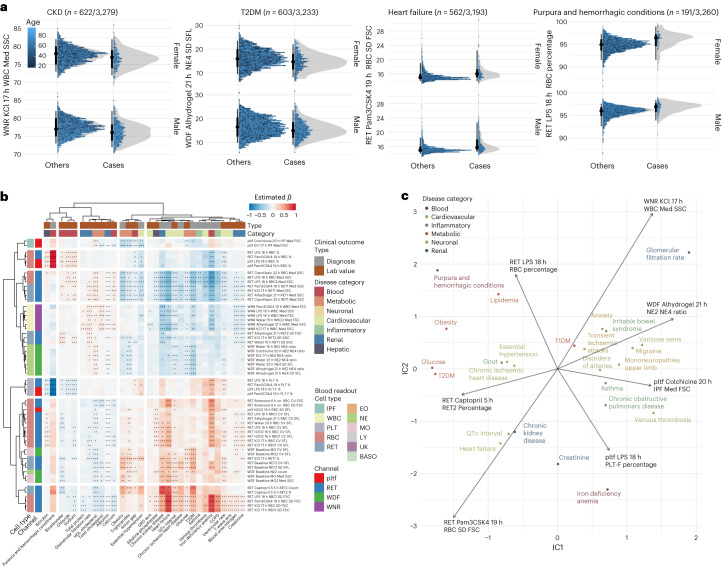
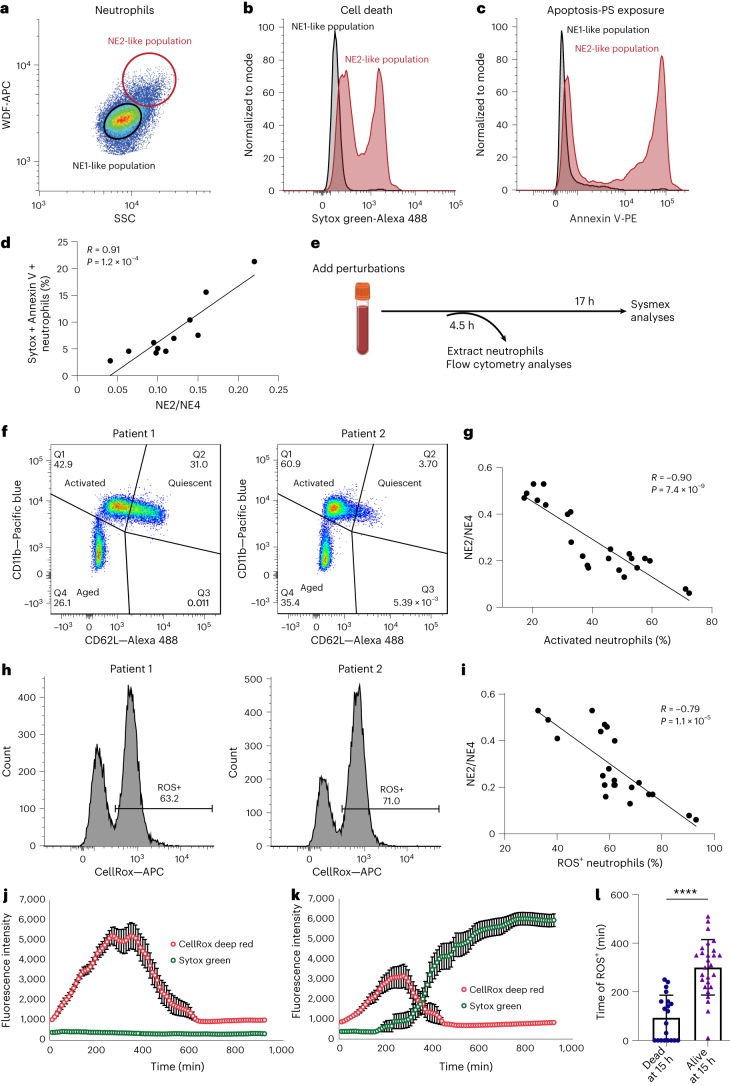
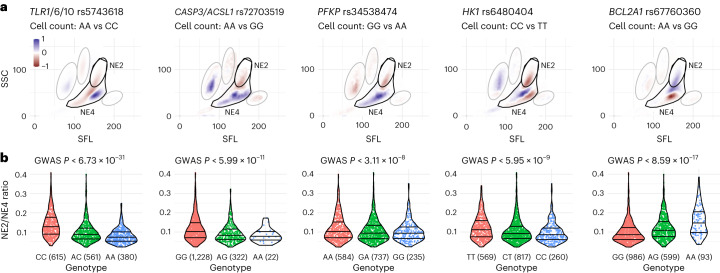
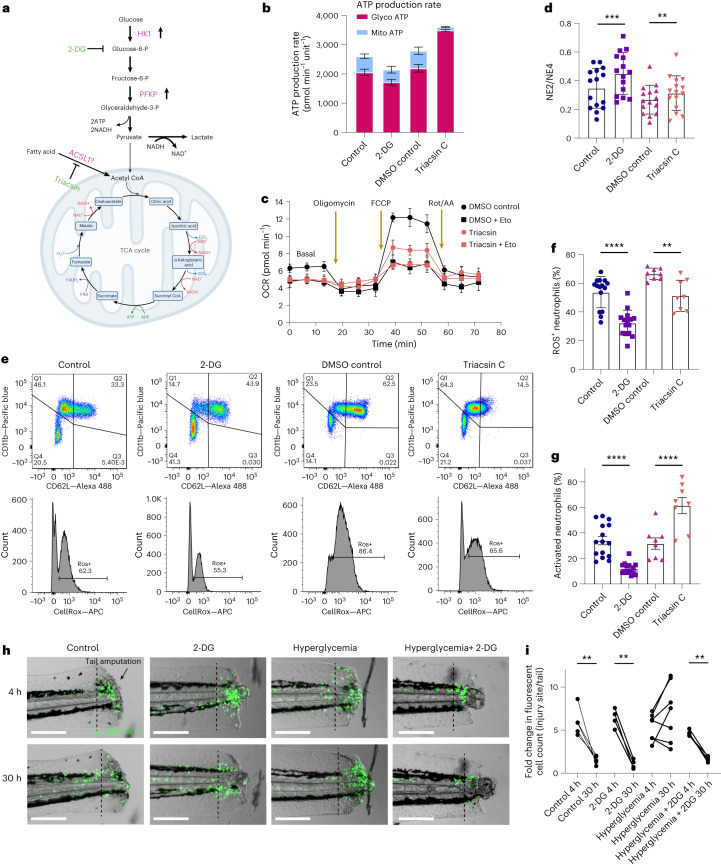
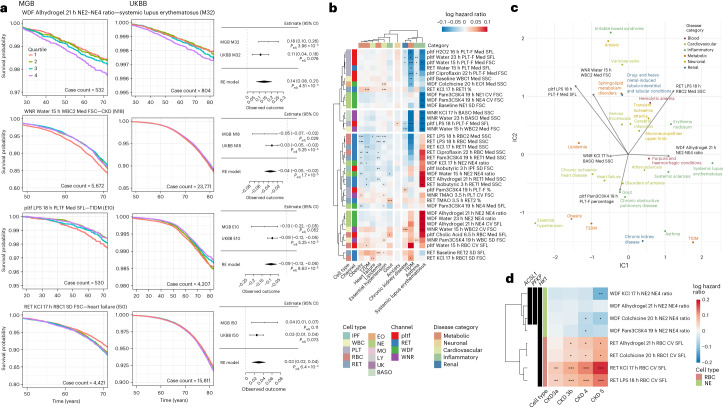
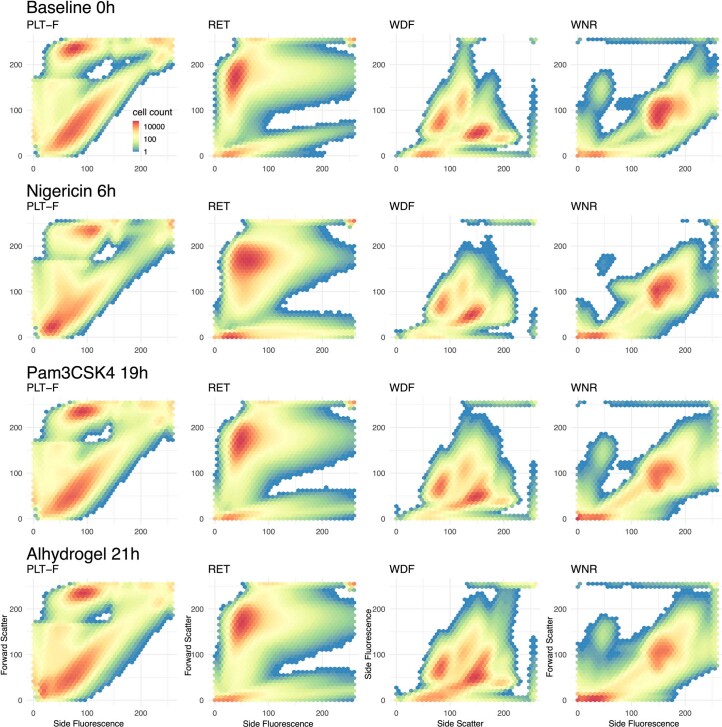
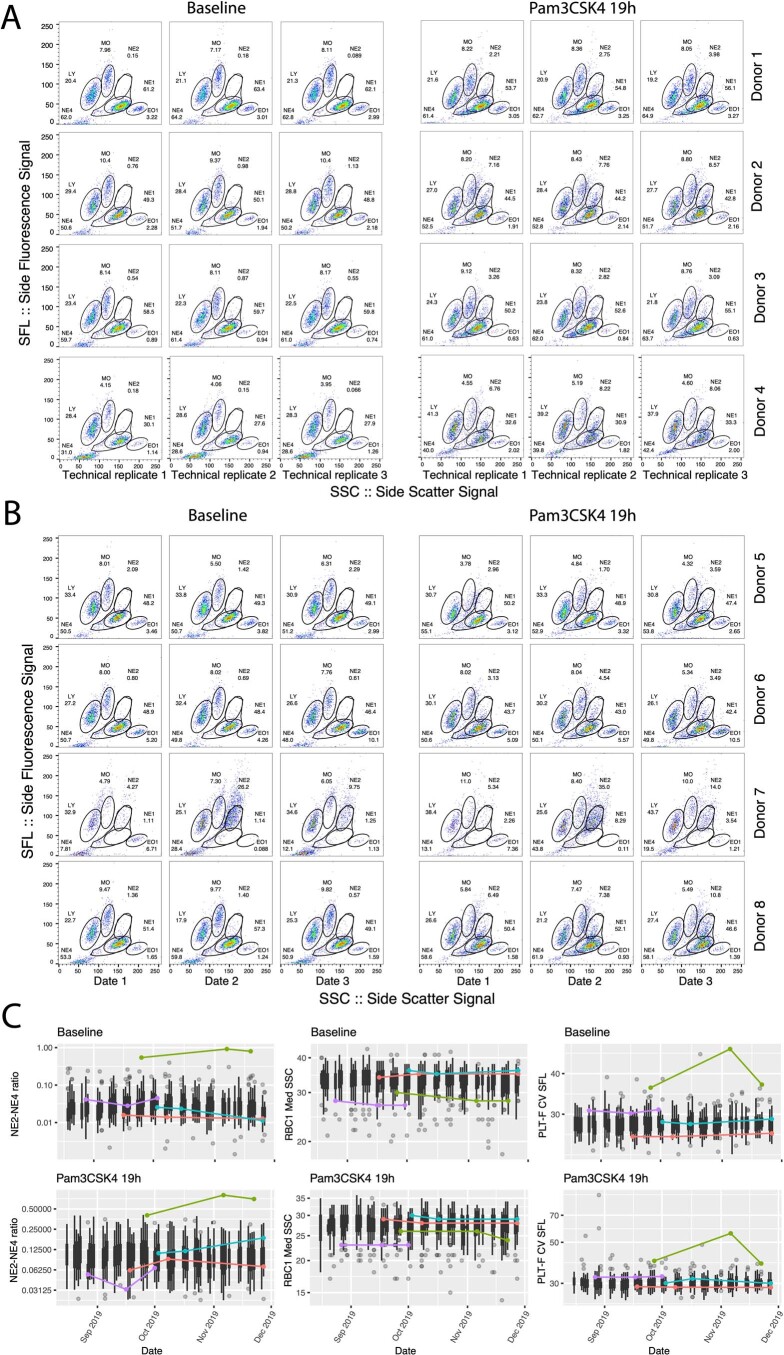
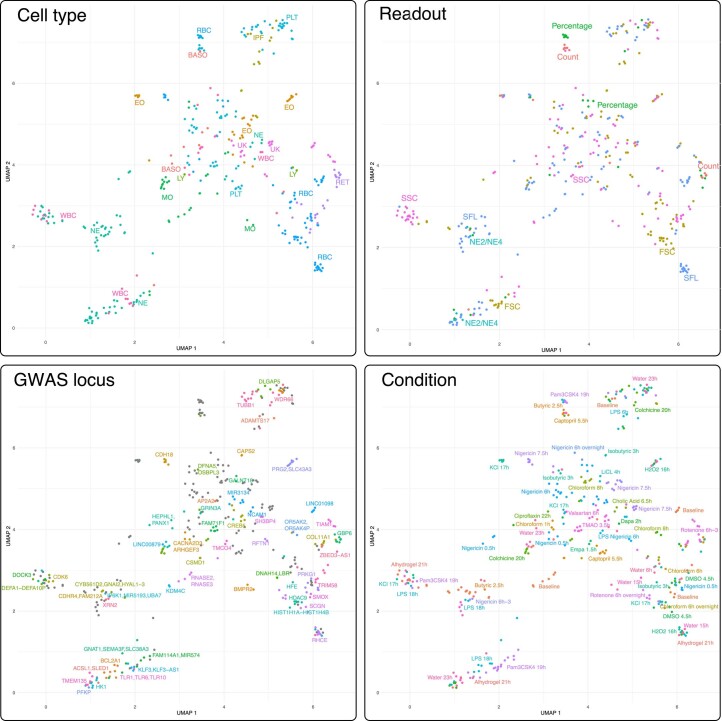
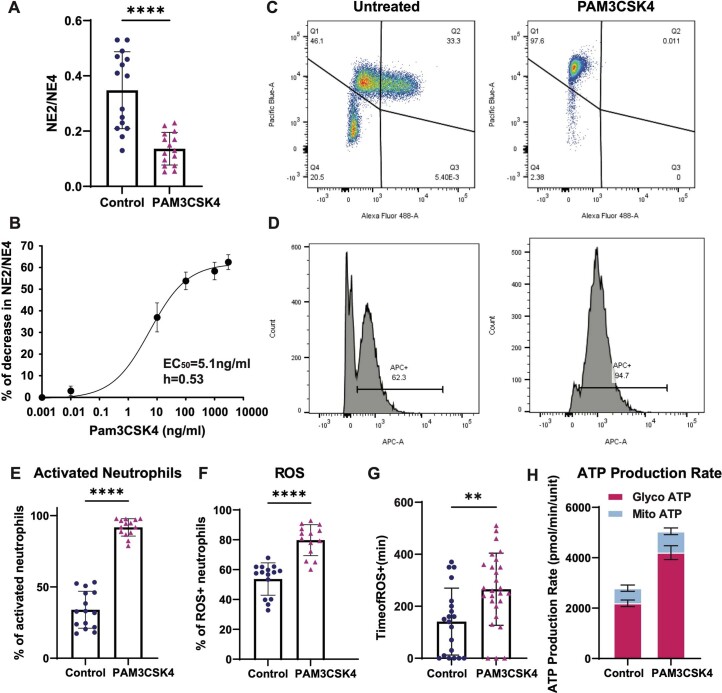
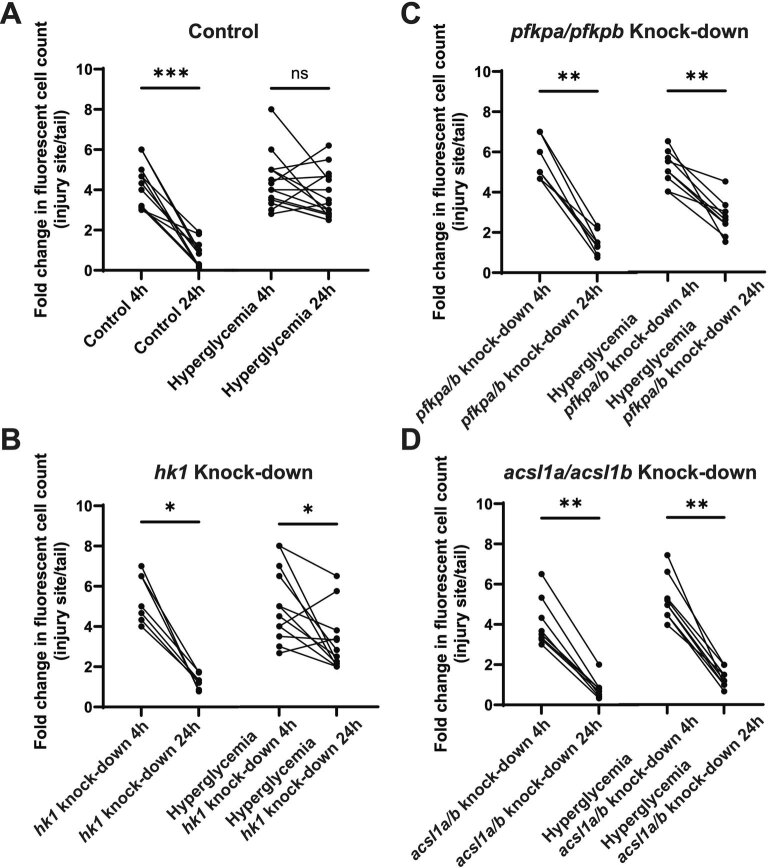
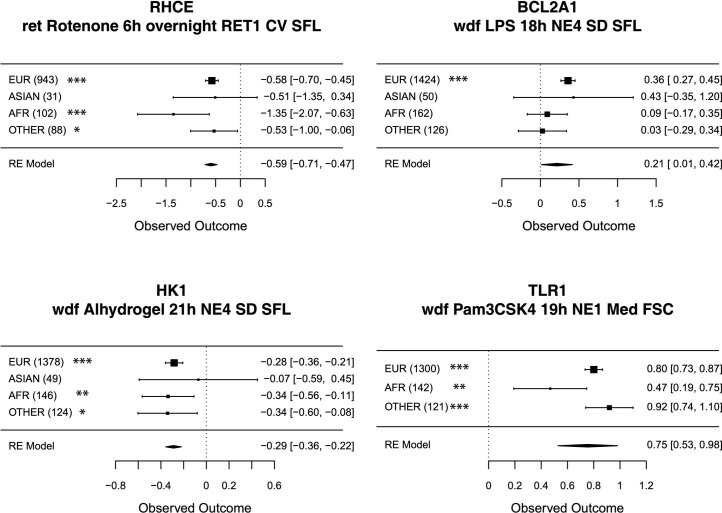
Although genome-wide association studies (GWAS) have successfully linked genetic risk loci to various disorders, identifying underlying cellular biological mechanisms remains challenging due to the complex nature of common diseases. We established a framework using human peripheral blood cells, physical, chemical and pharmacological perturbations, and flow cytometry-based functional readouts to reveal latent cellular processes and performed GWAS based on these evoked traits in up to 2,600 individuals. We identified 119 genomic loci implicating 96 genes associated with these cellular responses and discovered associations between evoked blood phenotypes and subsets of common diseases. We found a population of pro-inflammatory anti-apoptotic neutrophils prevalent in individuals with specific subsets of cardiometabolic disease. Multigenic models based on this trait predicted the risk of developing chronic kidney disease in type 2 diabetes patients. By expanding the phenotypic space for human genetic studies, we could identify variants associated with large effect response differences, stratify patients and efficiently characterize the underlying biology.
© 2023. The Author(s).
Conflict of interest statement
R.C.D. was supported by grants from the National Institutes of Health and the American Heart Association (One Brave Idea, Apple Heart and Movement Study) and is a cofounder of Atman Health. C.A.M. is supported by grants from the National Institutes of Health and the American Heart Association (One Brave Idea, Apple Heart and Movement Study); is a consultant for Bayer, Biosymetrics, Clarify Health, Dewpoint Therapeutics, Dinaqor, Dr. Evidence, Foresite Labs, Insmed, Pfizer and Purpose Life Sciences; and is a cofounder of Atman Health. R.C.D., M.H., C.A.M., S.V. and W.Z. are co-inventors on patents related to this work, and C.A.M., R.C.D., M.H. and W.Z. hold equity in Tanaist. All other authors report no competing interests.
Figures
References
-
- Martínez-Jiménez F, et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer. 2020;20:555–572. - PubMed
-
- Green RH, et al. Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet. 2002;360:1715–1721. - PubMed
-
- Gertz MA, Dispenzieri A. Systemic amyloidosis recognition, prognosis, and therapy. JAMA. 2020;324:79–89. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
