

The role of APOBEC3B in lung tumor evolution and targeted cancer therapy resistance
- PMID: 38049664
- PMCID: PMC10786726
- DOI: 10.1038/s41588-023-01592-8
The role of APOBEC3B in lung tumor evolution and targeted cancer therapy resistance
Abstract
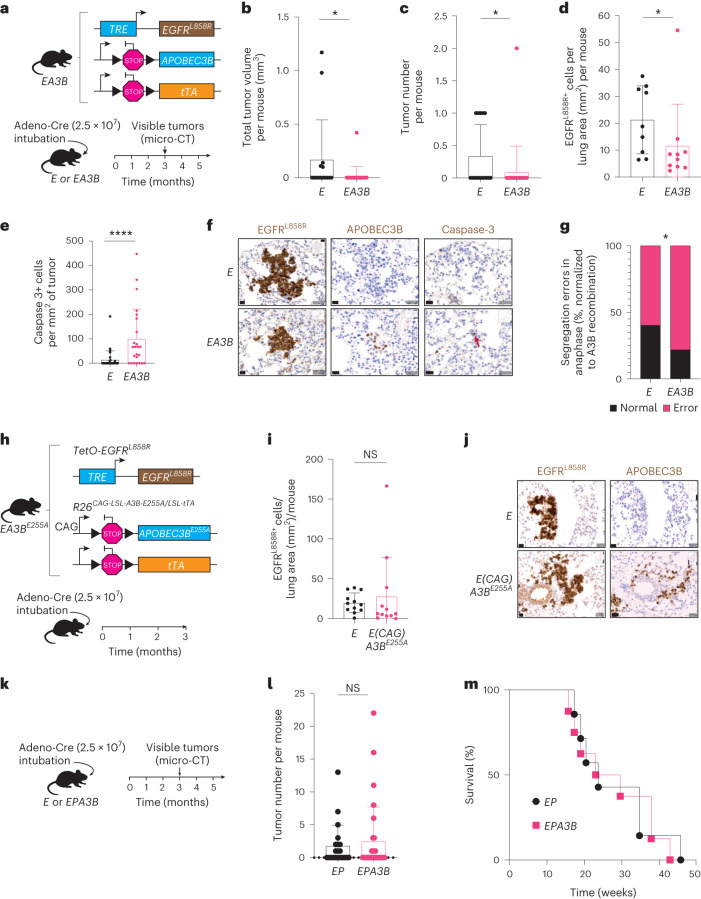
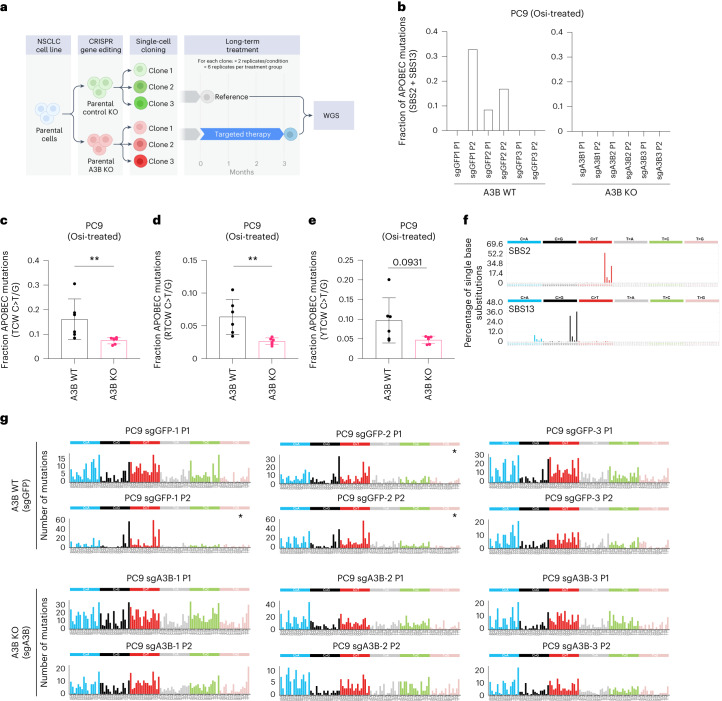
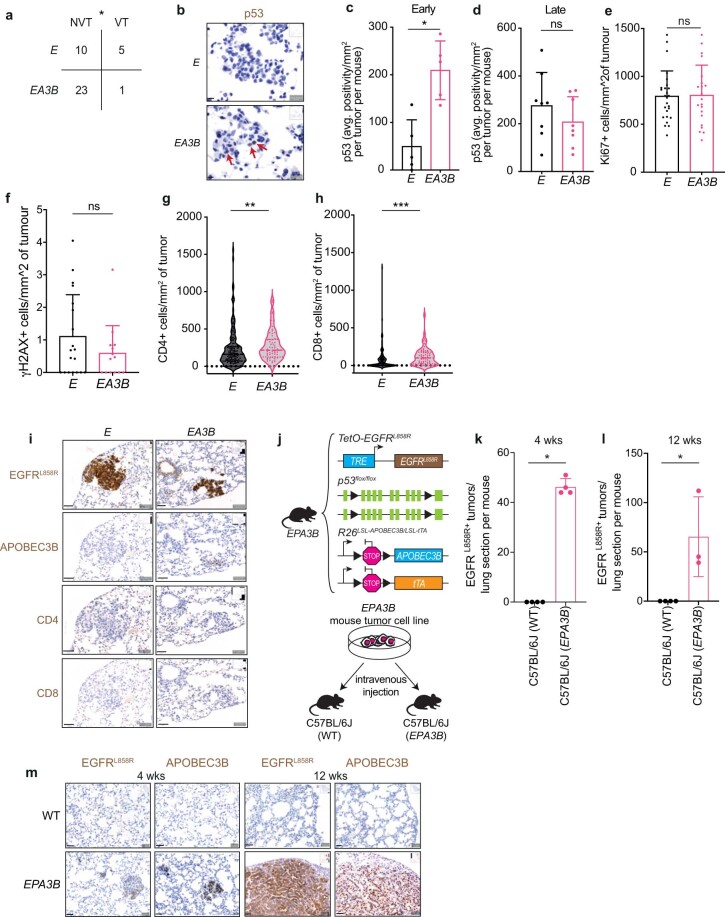
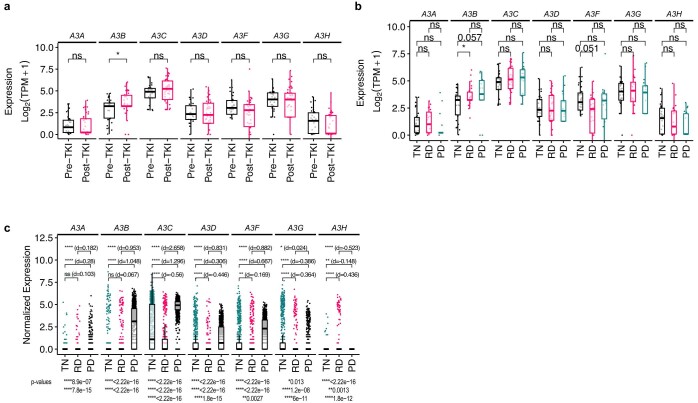
In this study, the impact of the apolipoprotein B mRNA-editing catalytic subunit-like (APOBEC) enzyme APOBEC3B (A3B) on epidermal growth factor receptor (EGFR)-driven lung cancer was assessed. A3B expression in EGFR mutant (EGFRmut) non-small-cell lung cancer (NSCLC) mouse models constrained tumorigenesis, while A3B expression in tumors treated with EGFR-targeted cancer therapy was associated with treatment resistance. Analyses of human NSCLC models treated with EGFR-targeted therapy showed upregulation of A3B and revealed therapy-induced activation of nuclear factor kappa B (NF-κB) as an inducer of A3B expression. Significantly reduced viability was observed with A3B deficiency, and A3B was required for the enrichment of APOBEC mutation signatures, in targeted therapy-treated human NSCLC preclinical models. Upregulation of A3B was confirmed in patients with NSCLC treated with EGFR-targeted therapy. This study uncovers the multifaceted roles of A3B in NSCLC and identifies A3B as a potential target for more durable responses to targeted cancer therapy.
© 2023. The Author(s).
Conflict of interest statement
T.G.B. is an advisor to Novartis, AstraZeneca, Revolution Medicines, Array/Pfizer, Springworks, Strategia, Relay, Jazz, Rain, Engine, Granule Therapeutics and EcoR1 and receives research funding from Novartis and Revolution Medicines, Kinnate, Verastem and Strategia. N.I.V. served on an advisory board for Sanofi Genzyme. C.S. acknowledges grants from AstraZeneca, Boehringer-Ingelheim, Bristol Myers Squibb, Pfizer, Roche-Ventana, Invitae (previously Archer Dx—collaboration in minimal RD sequencing technologies), Ono Pharmaceutical, and Personalis. He is the chief investigator for the AZ MeRmaiD 1 and 2 clinical trials and is the Steering Committee Chair. He is also co-chief investigator of the NHS Galleri trial funded by GRAIL and a paid member of GRAIL’s Scientific Advisory Board (SAB). He receives consultant fees from Achilles Therapeutics (also an SAB member), Bicycle Therapeutics (also an SAB member), Genentech, Medicxi, China Innovation Center of Roche (CICoR) formerly Roche Innovation Center—Shanghai, Metabomed (until July 2022), Relay Therapeutics and the Sarah Cannon Research Institute. C.S. has received honoraria from Amgen, AstraZeneca, Bristol Myers Squibb, GlaxoSmithKline, Illumina, MSD, Novartis, Pfizer and Roche-Ventana; has previously held stock options in Apogen Biotechnologies and GRAIL; currently has stock options in Epic Bioscience and Bicycle Therapeutics and has stock options and is a cofounder of Achilles Therapeutics. C.S. declares a patent application (PCT/US2017/028013) for methods to lung cancer; targeting neoantigens (PCT/EP2016/059401); identifying patent response to immune checkpoint blockade (PCT/EP2016/071471), determining HLA LOH (PCT/GB2018/052004); predicting survival rates of patients with cancer (PCT/GB2020/050221), identifying patients who respond to cancer treatment (PCT/GB2018/051912); methods for lung cancer detection (US20190106751A1). He is an inventor on a European patent application (PCT/GB2017/053289) relating to assay technology to detect tumor recurrence. This patent has been licensed to a commercial entity, and under their terms of employment, C.S. is due a revenue share of any revenue generated from such license(s). E.M.V.A. is a consultant for Tango Therapeutics, Genome Medical, Invitae, Enara Bio, Janssen, Manifold Bio, Monte Rosa; receives research funding from Novartis, BMS; has equity in Tango Therapeutics, Genome Medical, Syapse, Enara Bio, Manifold Bio, Microsoft and Monte Rosa; has received travel reimbursement from Roche/Genentech and own institutional patents filed on chromatin mutations and immunotherapy response, and methods for clinical interpretation. C.E.M. is on the advisory board of Genentech; receives honoraria from Novartis, Guardant, Research and receives funding from Novartis, Revolution Medicines. C.M.B. is a consultant for Amgen, Foundation Medicine, Blueprint Medicines and Revolution Medicines; receives research funding from Novartis, AstraZeneca and Takeda and receives institutional research funding from Mirati, Spectrum, MedImmune and Roche. J.S.R.-F. reports receiving personal/consultancy fees from Goldman Sachs, Bain Capital, REPARE Therapeutics, Saga Diagnostics and Paige.AI, membership of the SAB of VolitionRx, REPARE Therapeutics and Paige.AI, membership of the Board of Directors (BOD) of Grupo Oncoclinicas, and ad hoc SAB of Astrazeneca, Merck, Daiichi Sankyo, Roche Tissue Diagnostics and Personalis, outside the scope of this study. H.Y. receives consulting fees from AstraZeneca, Daiichi, Taiho, Janssen, AbbVie, Blueprint, Black Diamond Research funding to my institution from AstraZeneca, Daiichi, Cullinan, Janssen, Blueprint, Black Diamond, Novartis, Pfizer, ERASCA. S.F.B. owns equity in, receives compensation from, serves as a consultant for and serves on the SAB and BOD of Volastra Therapeutics. He serves on the scientific advisory board of Meliora Therapeutics. M.J.-H. has consulted for, and is a member of, the Achilles Therapeutics Scientific Advisory Board and Steering Committee; has received speaker honoraria from Pfizer, Astex Pharmaceuticals, Oslo Cancer Cluster and Bristol Myers Squibb and is listed as a co-inventor on a European patent application relating to methods to detect lung cancer (PCT/US2017/028013). This patent has been licensed to commercial entities and, under terms of employment, M.J.-H. is due a share of any revenue generated from such license(s). The other authors have no competing interests to declare.
Figures














References
-
- Swanton C, McGranahan N, Starrett GJ, Harris RS. APOBEC enzymes: mutagenic fuel for cancer evolution and heterogeneity. Cancer Discov. 2015;5:704–712. doi: 10.1158/2159-8290.CD-15-0344. - DOI - PMC - PubMed
-
- Venkatesan S, et al. Induction of APOBEC3 exacerbates DNA replication stress and chromosomal instability in early breast and lung cancer evolution. Cancer Discov. 2021;11:2456–2473. doi: 10.1158/2159-8290.CD-20-0725. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
- T32 GM140936/GM/NIGMS NIH HHS/United States
- 21999/CRUK_/Cancer Research UK/United Kingdom
- U01 CA217882/CA/NCI NIH HHS/United States
- R01 CA227807/CA/NCI NIH HHS/United States
- T32 HL007185/HL/NHLBI NIH HHS/United States
- R01 CA211052/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- CC2041/WT_/Wellcome Trust/United Kingdom
- R01 CA204302/CA/NCI NIH HHS/United States
- U54 CA224081/CA/NCI NIH HHS/United States
- 17786/CRUK_/Cancer Research UK/United Kingdom
- P01 CA234228/CA/NCI NIH HHS/United States
- 30025/CRUK_/Cancer Research UK/United Kingdom
- P50 CA247749/CA/NCI NIH HHS/United States
- R01 CA264078/CA/NCI NIH HHS/United States
- FC001169/WT_/Wellcome Trust/United Kingdom
- R01 CA169338/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
