

Cyclic voltammetry and chronoamperometry: mechanistic tools for organic electrosynthesis
- PMID: 38050749
- PMCID: PMC10842901
- DOI: 10.1039/d2cs00706a
Cyclic voltammetry and chronoamperometry: mechanistic tools for organic electrosynthesis
Abstract
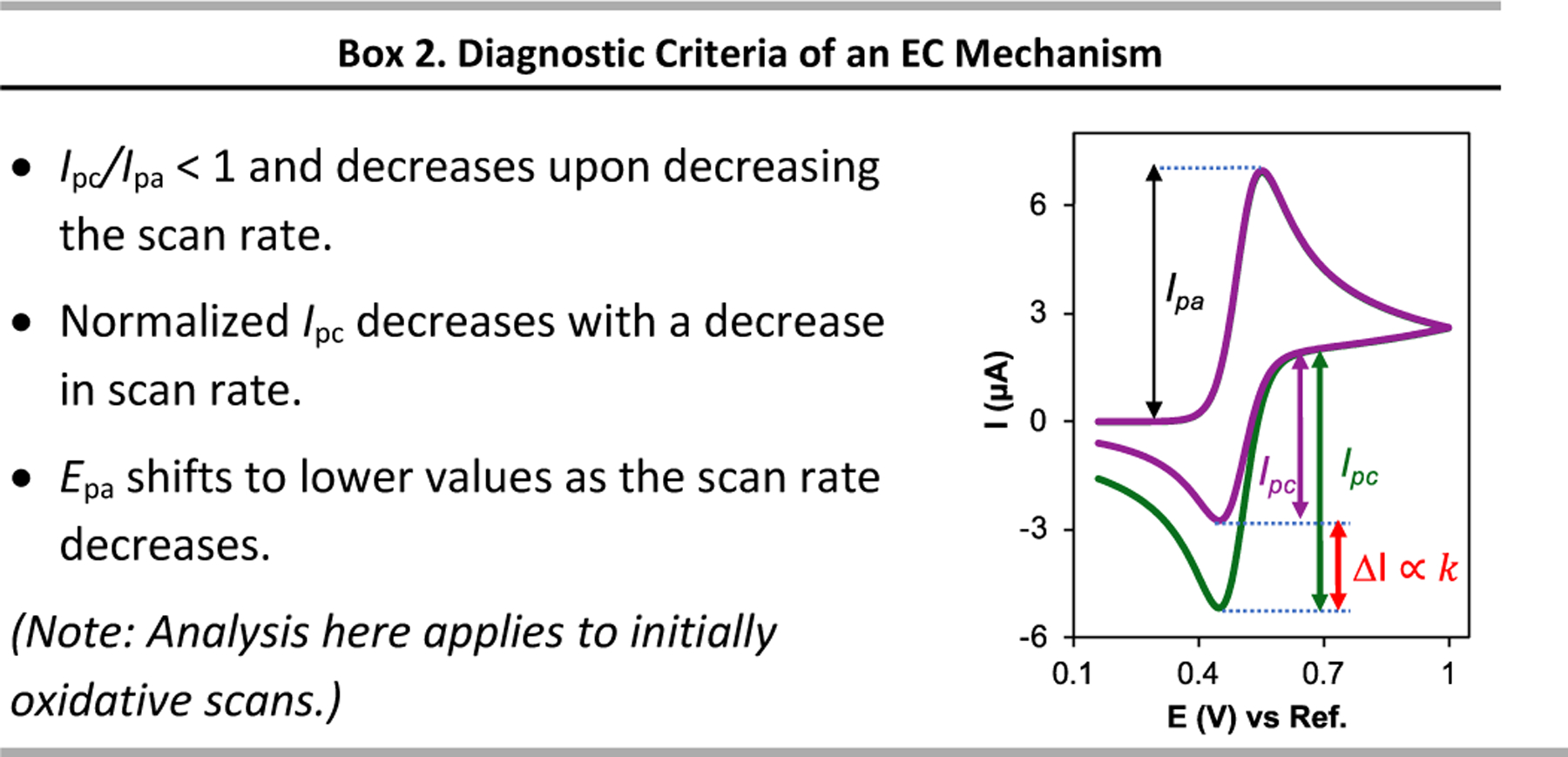
Electrochemical methods offer unique advantages for chemical synthesis, as the reaction selectivity may be controlled by tuning the applied potential or current. Similarly, measuring the current or potential during the reaction can provide valuable mechanistic insights into these reactions. The aim of this tutorial review is to explain the use of cyclic voltammetry and chronoamperometry to interrogate reaction mechanisms, optimize electrochemical reactions, or design new reactions. Fundamental principles of cyclic voltammetry and chronoamperometry experiments are presented together with the application of these techniques to probe (electro)chemical reactions. Several diagnostic criteria are noted for the use of cyclic voltammetry and chronoamperometry to analyze coupled electrochemical-chemical (EC) reactions, and a series of individual mechanistic studies are presented. Steady state voltammetric and amperometric measurements, using microelectrodes (ME) or rotating disk electrodes (RDE) provide a means to analyze concentrations of redox active species in bulk solution and offer a versatile strategy to conduct kinetic analysis or determine the species present during (electro)synthetic chemical reactions.
Conflict of interest statement
Conflicts of interest
There are no conflicts to declare.
Figures



























References
-
- Barek J and Zima J, Electroanalysis, 2003, 15, 467–472.
-
- McKenzie ECR, Hosseini S, Petro AGC, Rudman KK, Gerroll BHR, Mubarak MS, Baker LA and Little RD, Chem. Rev, 2022, 122, 3292–3335. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources