

Design and structural validation of peptide-drug conjugate ligands of the kappa-opioid receptor
- PMID: 38052802
- PMCID: PMC10698194
- DOI: 10.1038/s41467-023-43718-w
Design and structural validation of peptide-drug conjugate ligands of the kappa-opioid receptor
Abstract
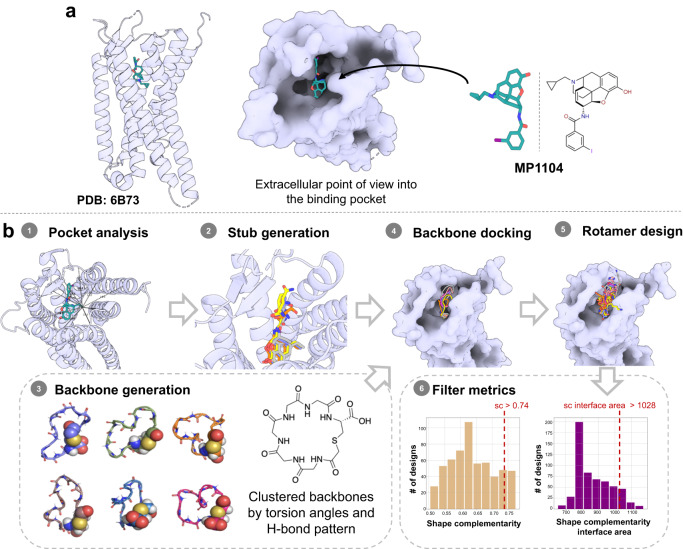
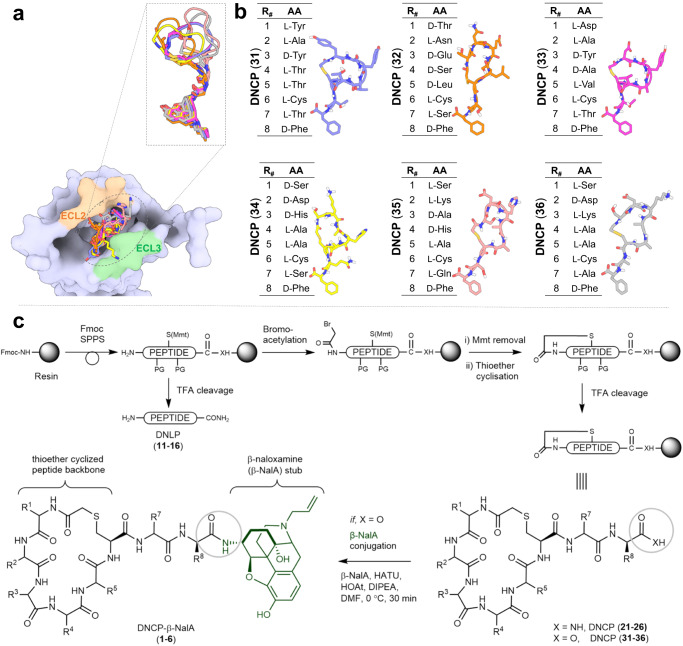
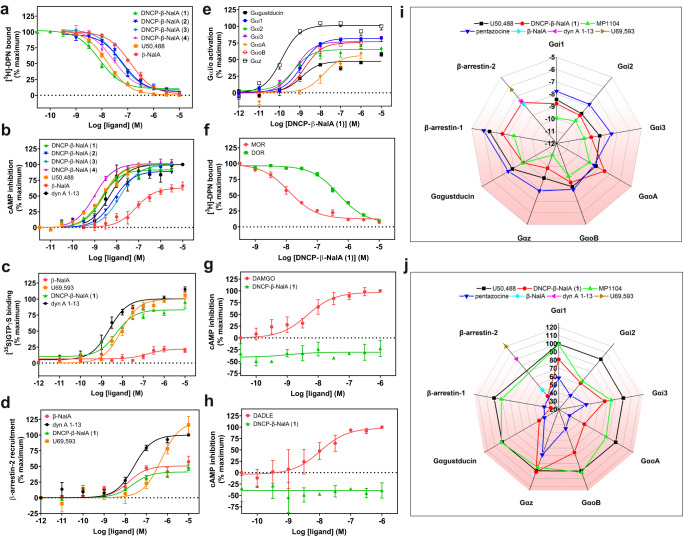
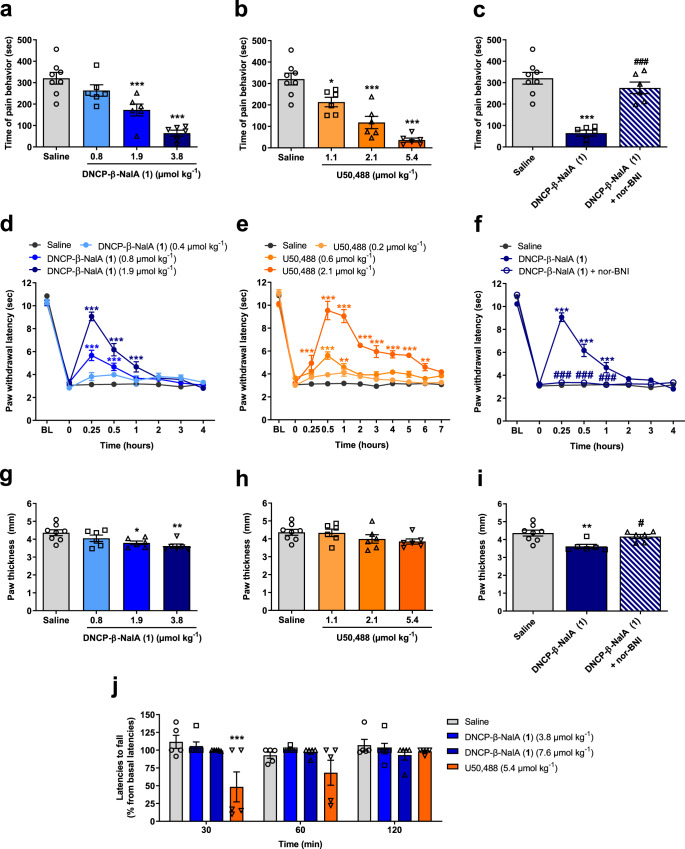
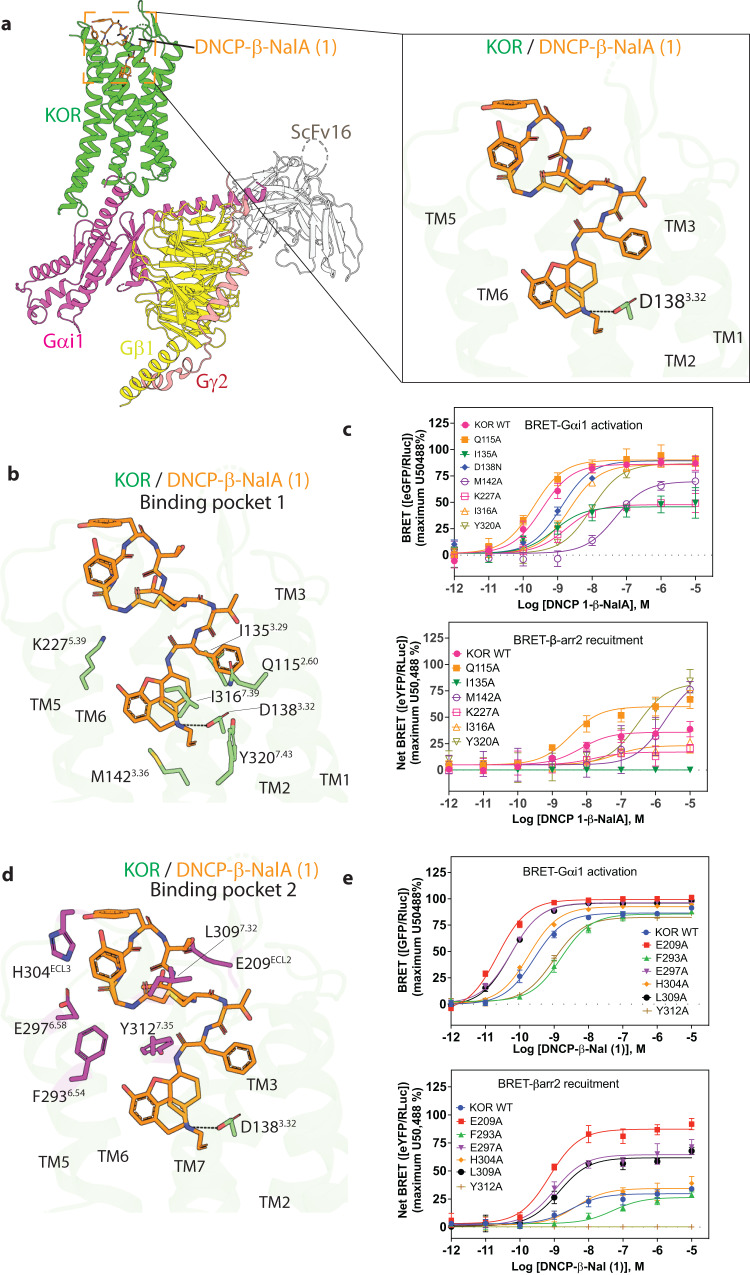
Despite the increasing number of GPCR structures and recent advances in peptide design, the development of efficient technologies allowing rational design of high-affinity peptide ligands for single GPCRs remains an unmet challenge. Here, we develop a computational approach for designing conjugates of lariat-shaped macrocyclized peptides and a small molecule opioid ligand. We demonstrate its feasibility by discovering chemical scaffolds for the kappa-opioid receptor (KOR) with desired pharmacological activities. The designed De Novo Cyclic Peptide (DNCP)-β-naloxamine (NalA) exhibit in vitro potent mixed KOR agonism/mu-opioid receptor (MOR) antagonism, nanomolar binding affinity, selectivity, and efficacy bias at KOR. Proof-of-concept in vivo efficacy studies demonstrate that DNCP-β-NalA(1) induces a potent KOR-mediated antinociception in male mice. The high-resolution cryo-EM structure (2.6 Å) of the DNCP-β-NalA-KOR-Gi1 complex and molecular dynamics simulations are harnessed to validate the computational design model. This reveals a network of residues in ECL2/3 and TM6/7 controlling the intrinsic efficacy of KOR. In general, our computational de novo platform overcomes extensive lead optimization encountered in ultra-large library docking and virtual small molecule screening campaigns and offers innovation for GPCR ligand discovery. This may drive the development of next-generation therapeutics for medical applications such as pain conditions.
© 2023. The Author(s).
Conflict of interest statement
S.M. is a co-founder of Sparian Biosciences. The other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
