

3D Enhancer-promoter networks provide predictive features for gene expression and coregulation in early embryonic lineages
- PMID: 38053013
- PMCID: PMC10897904
- DOI: 10.1038/s41594-023-01130-4
3D Enhancer-promoter networks provide predictive features for gene expression and coregulation in early embryonic lineages
Abstract
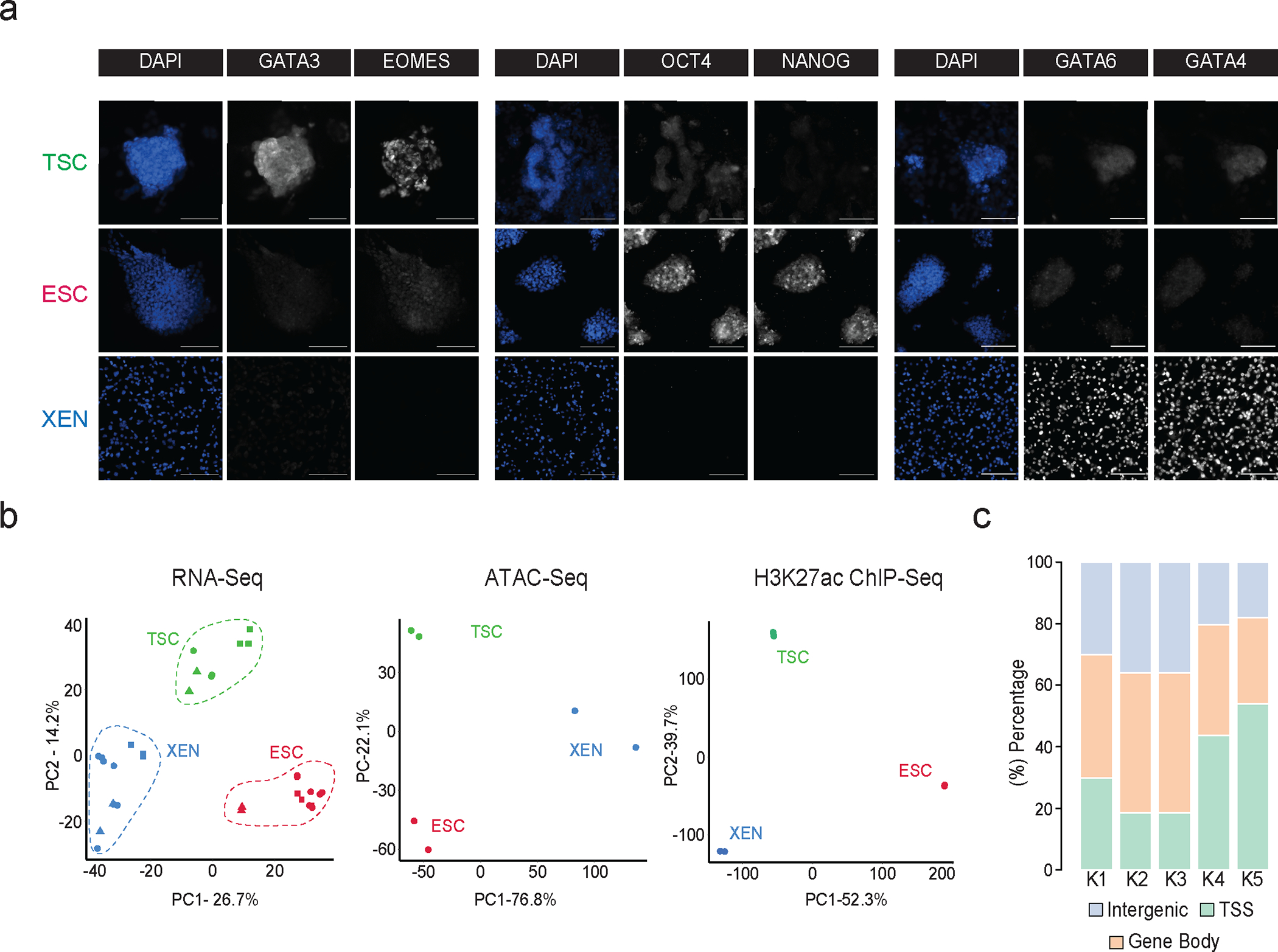
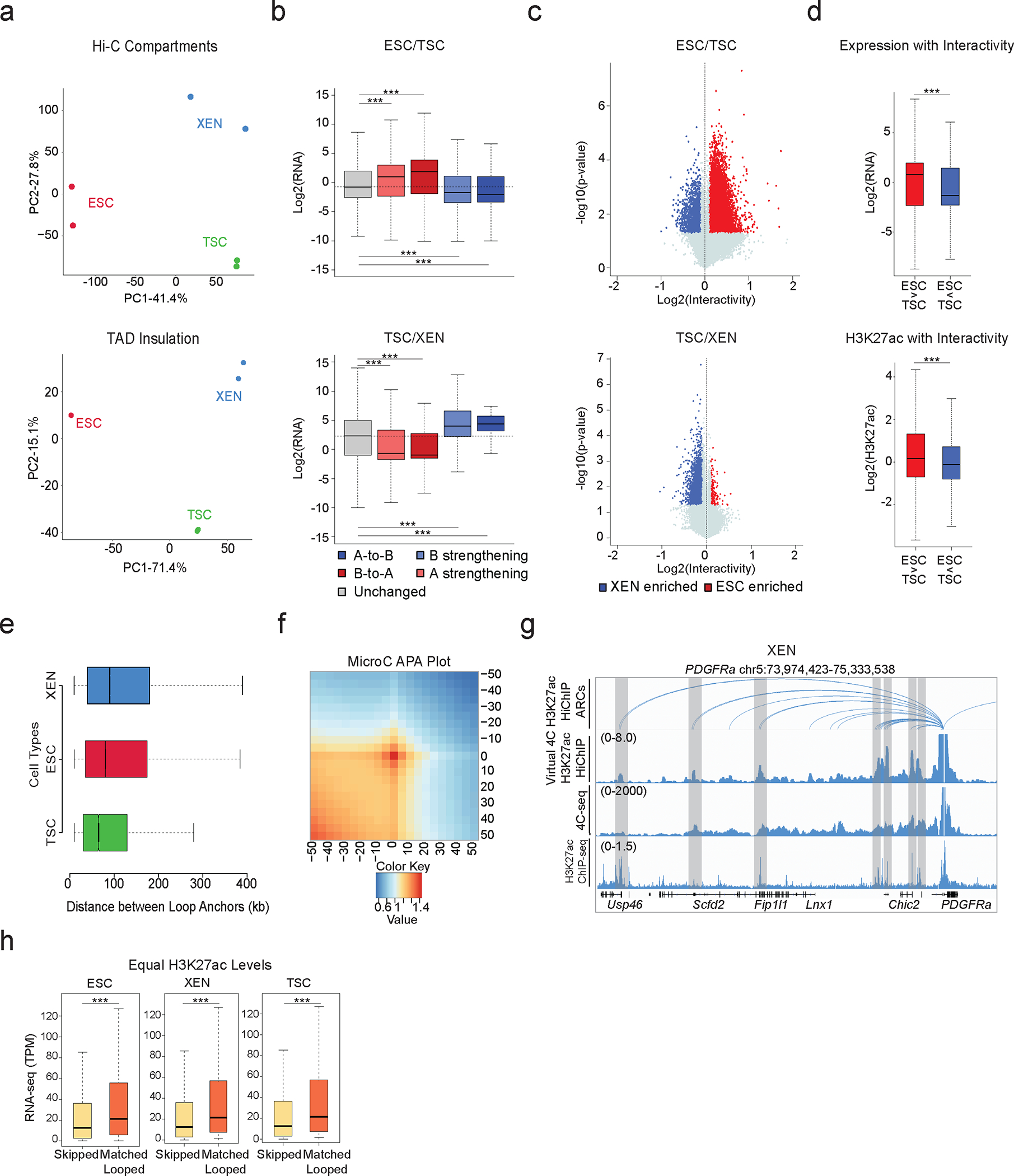
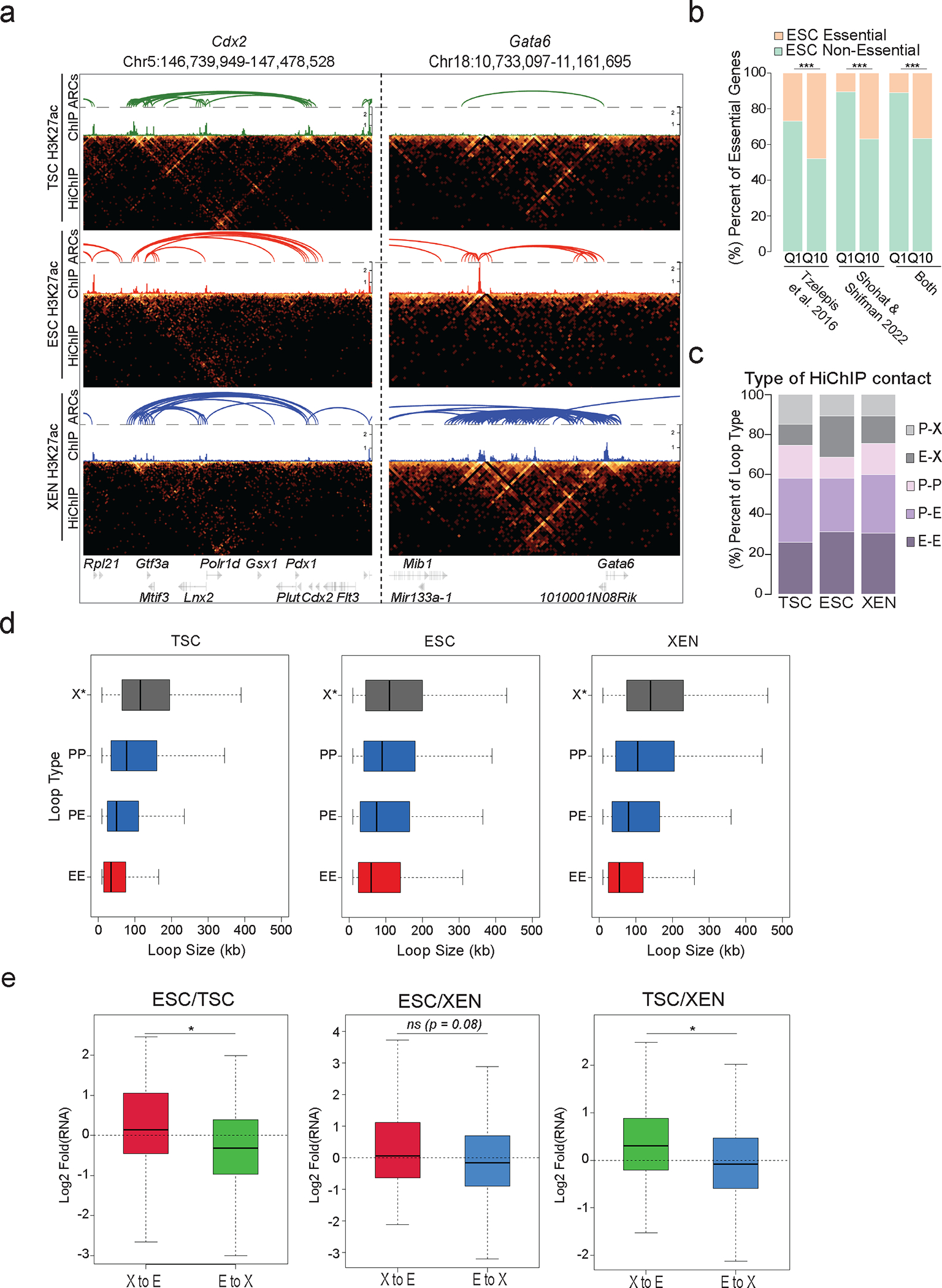
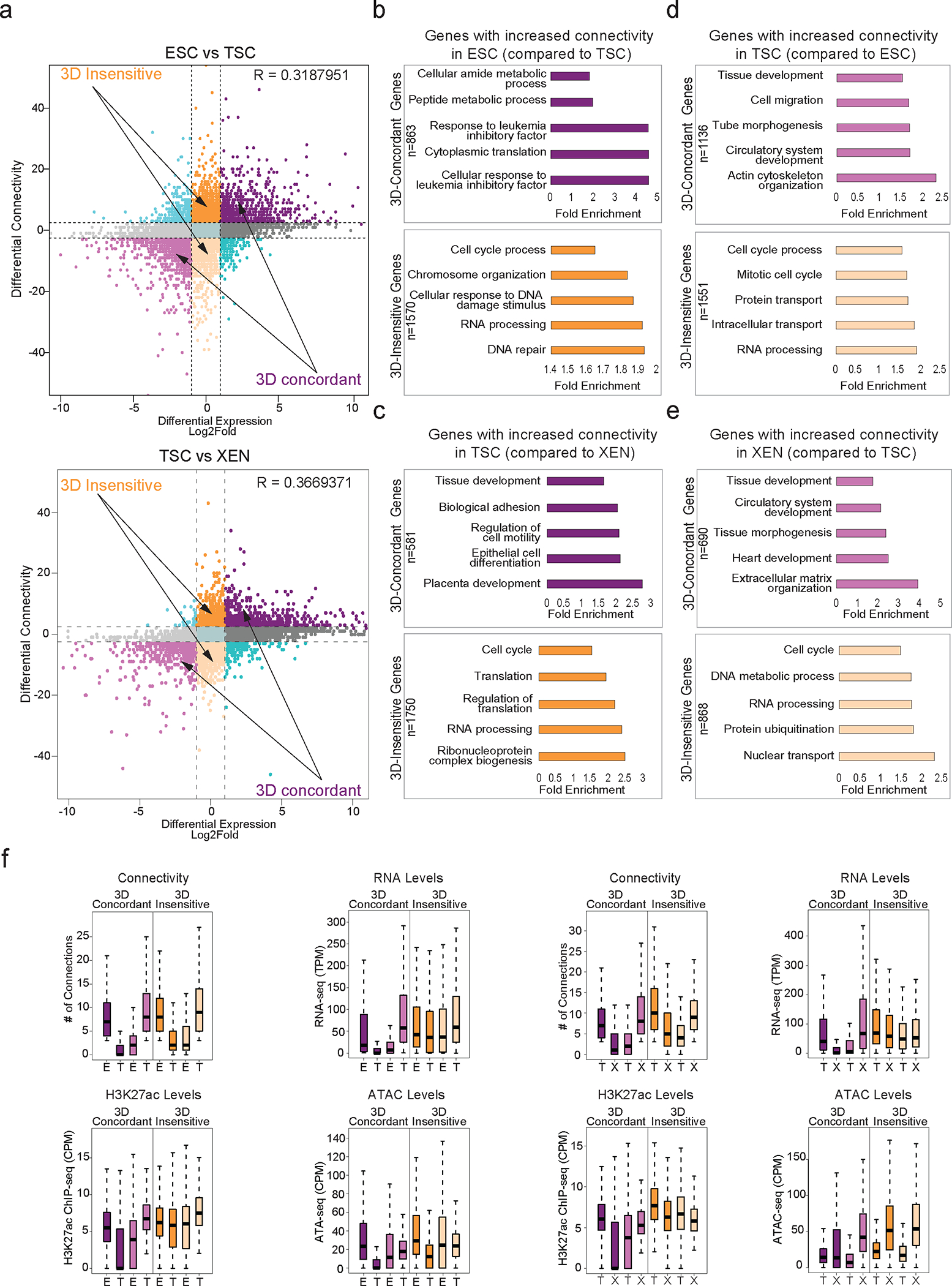
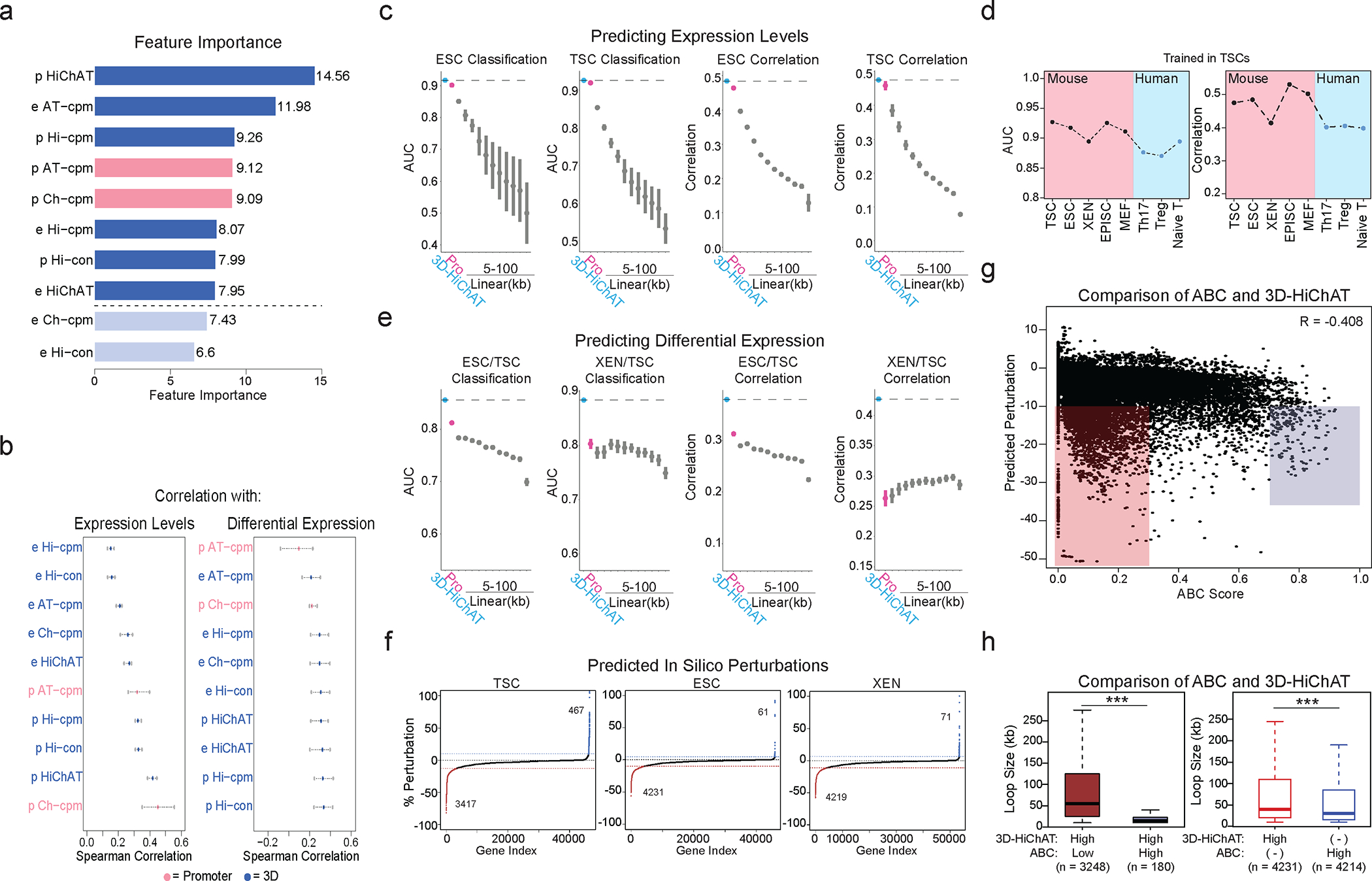
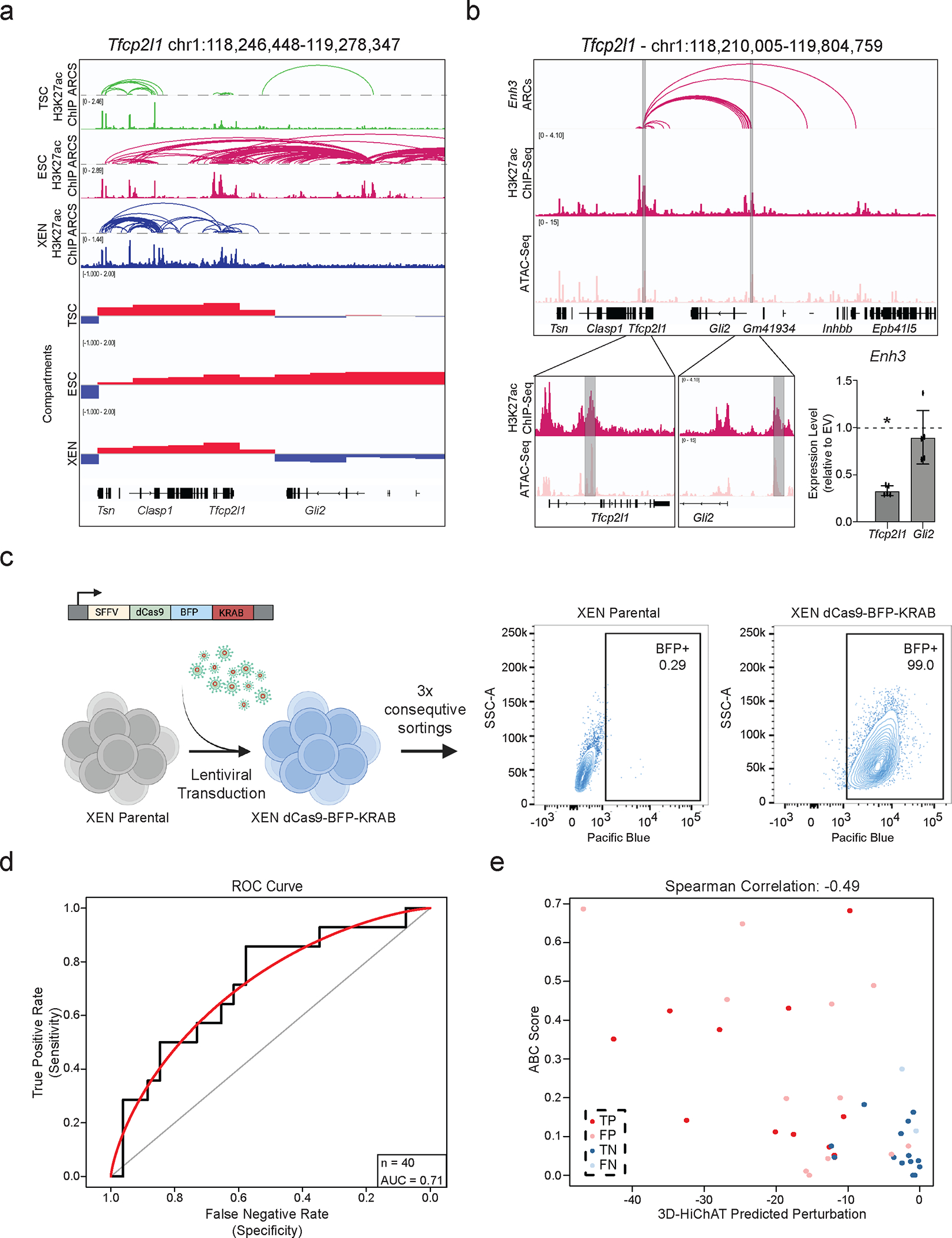
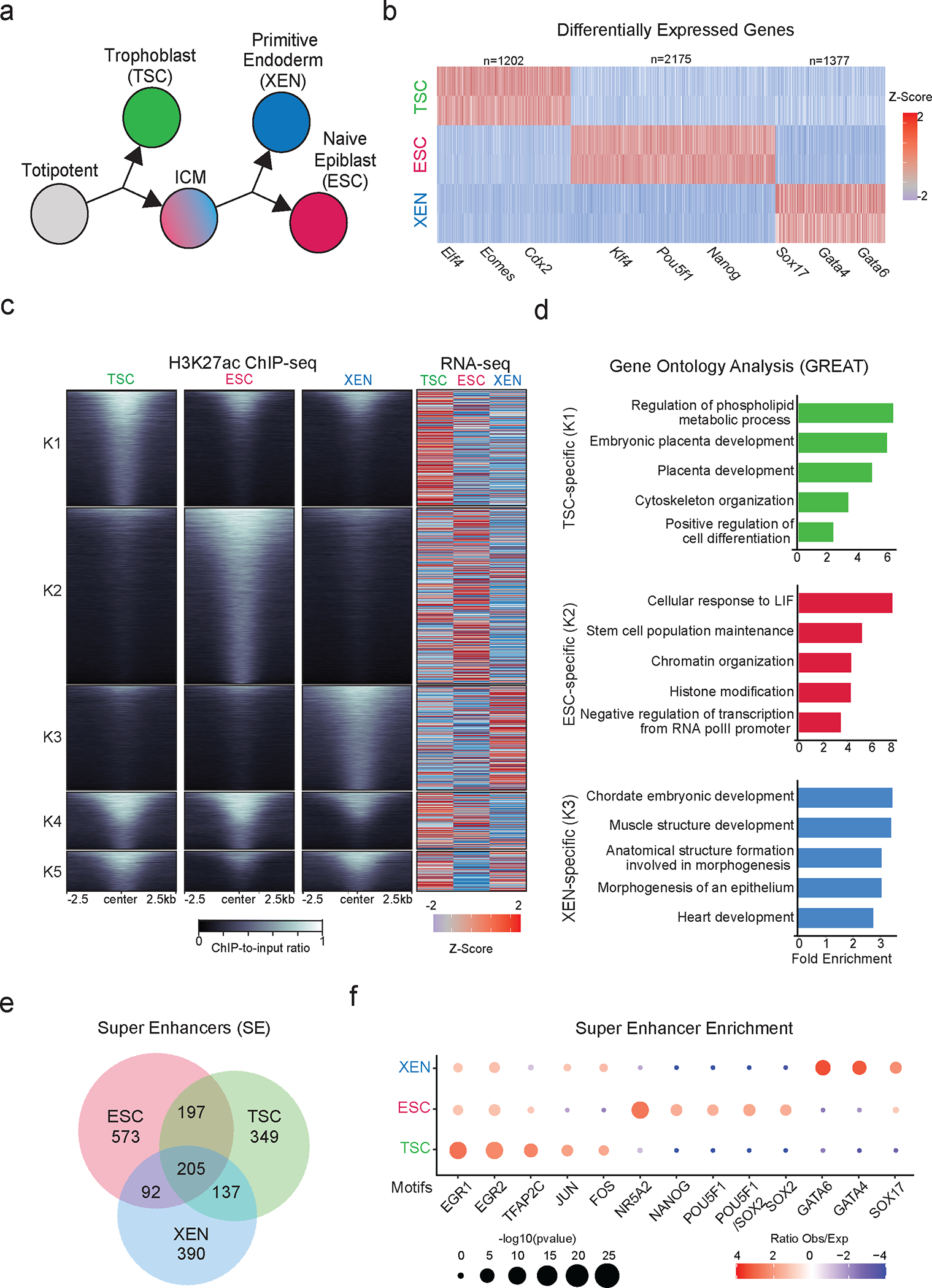
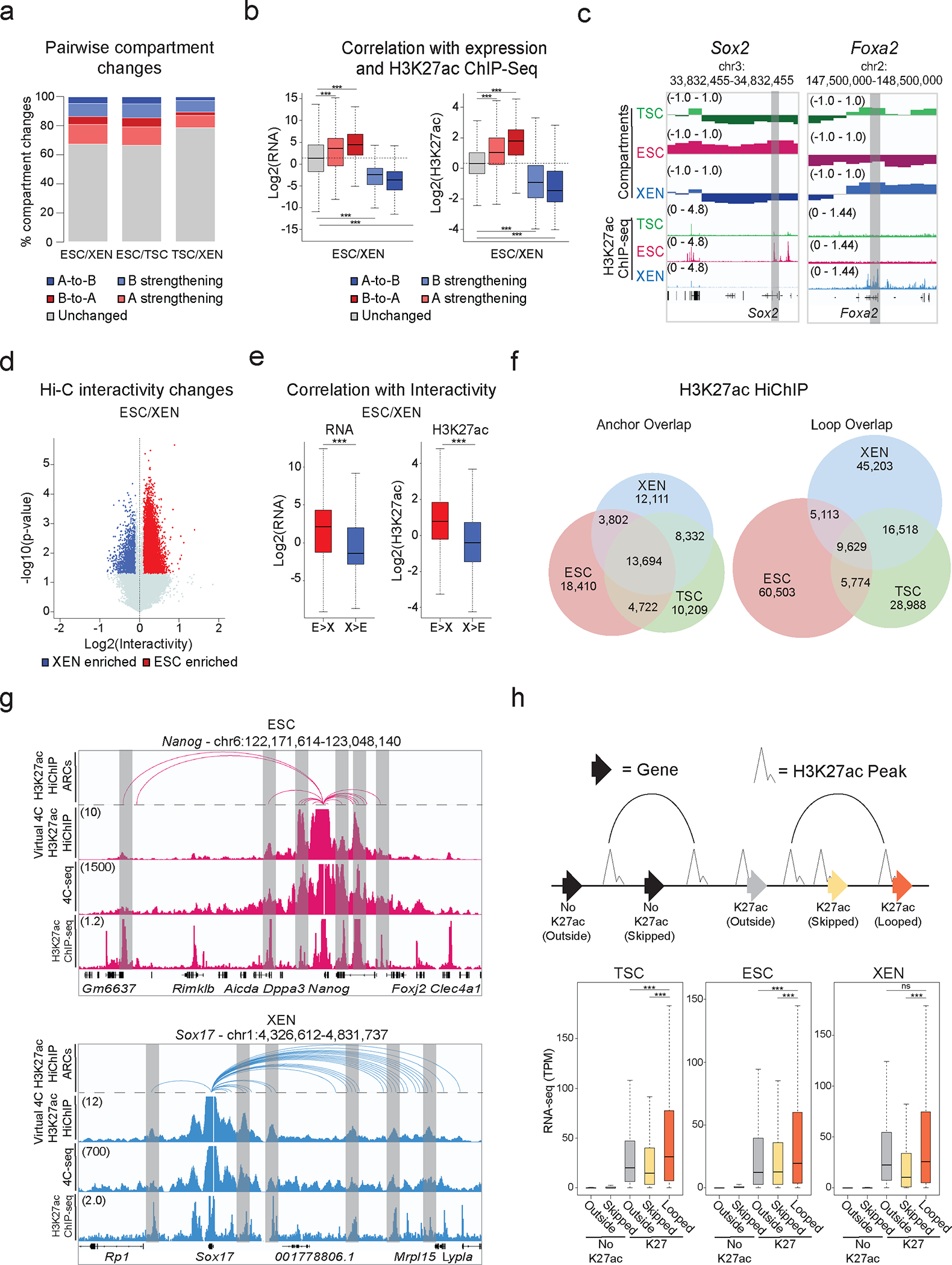
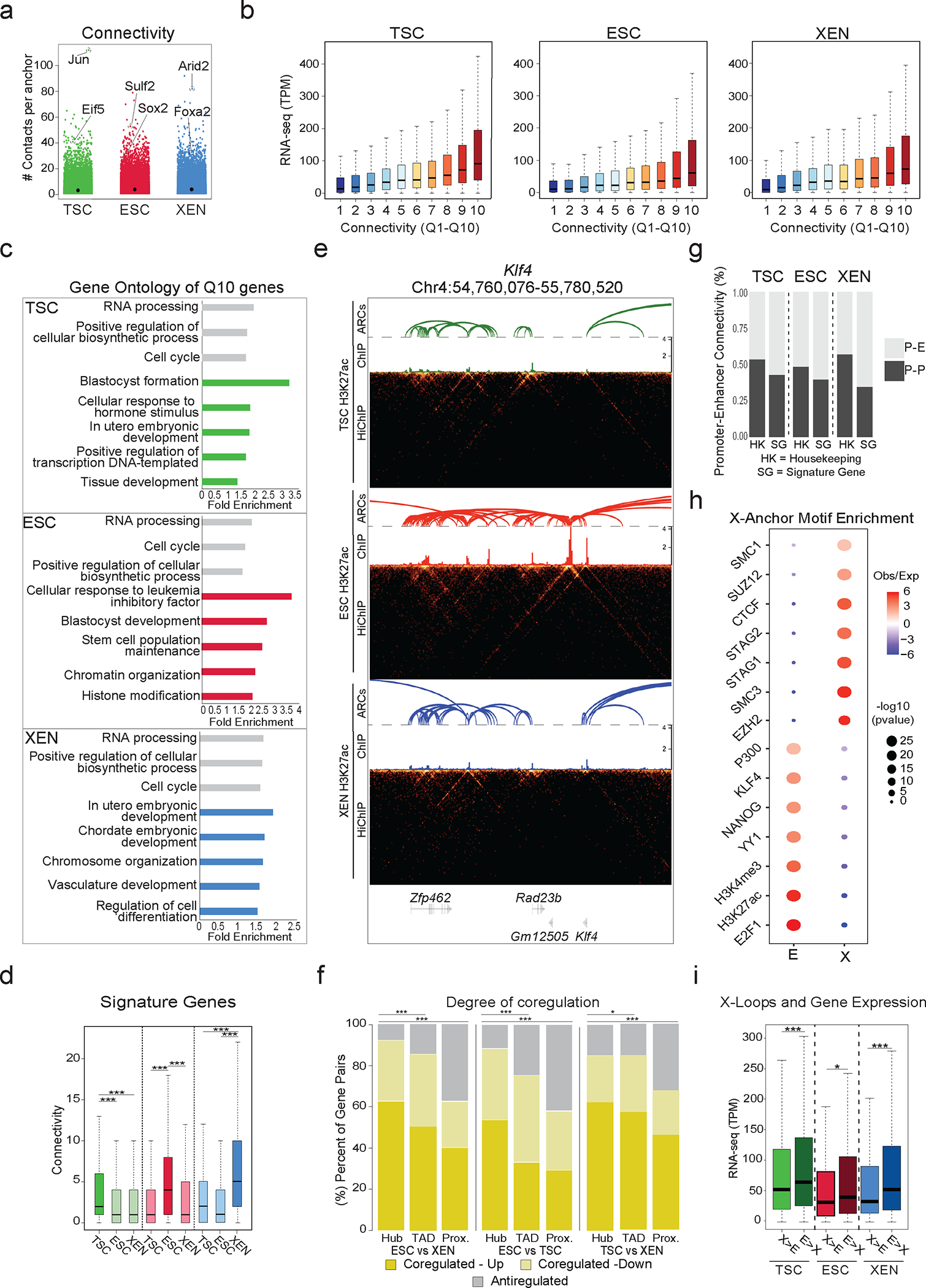
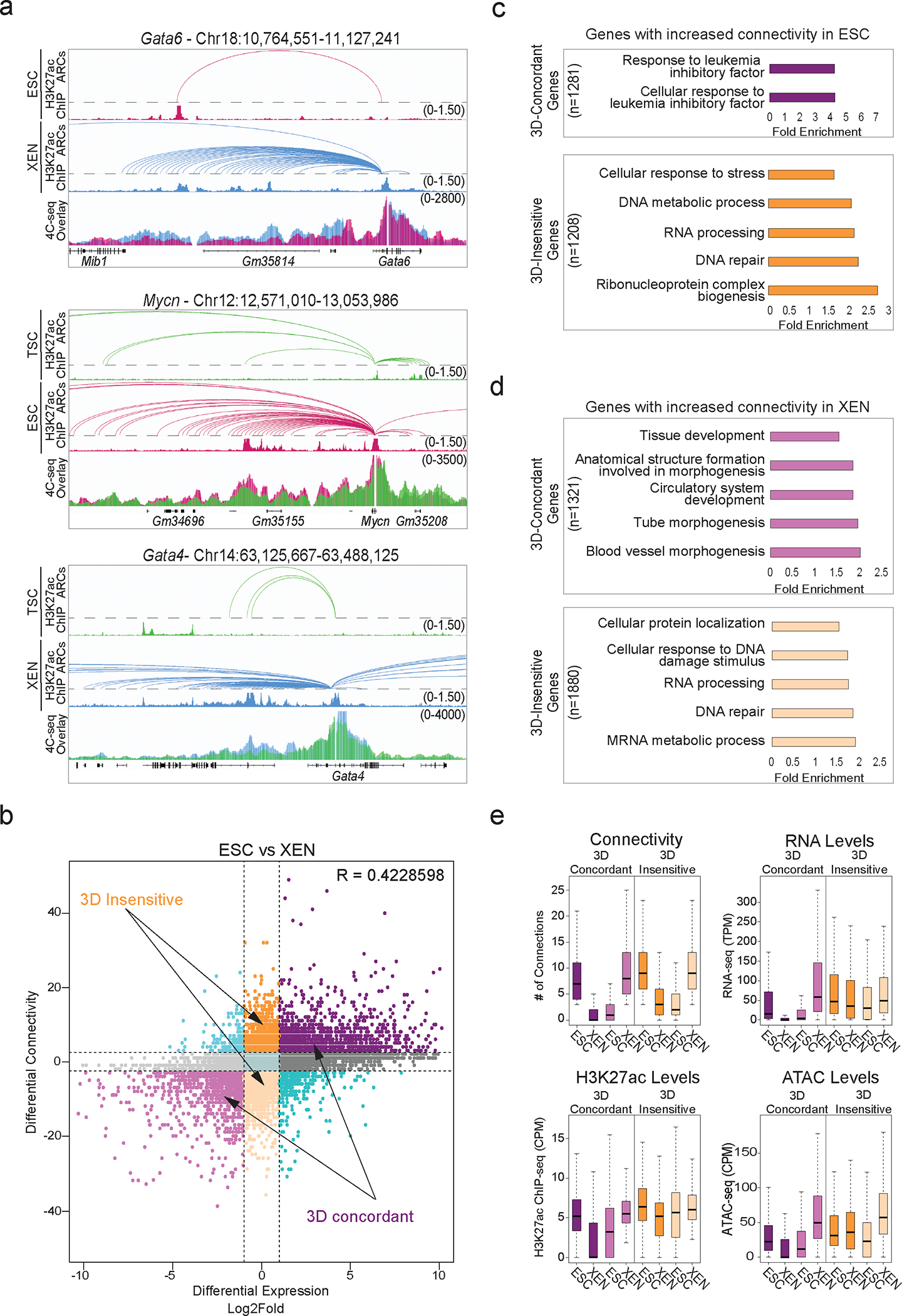
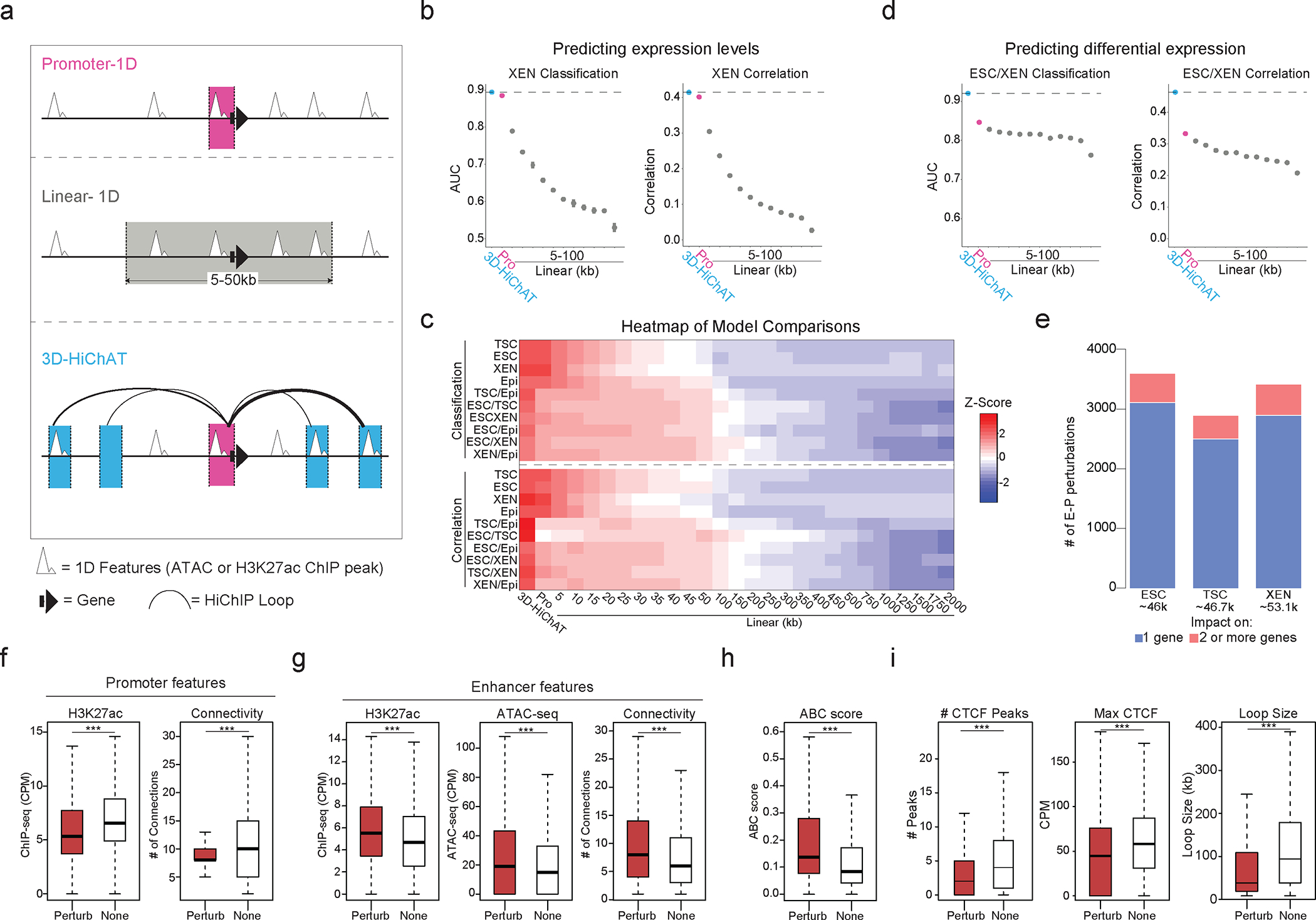
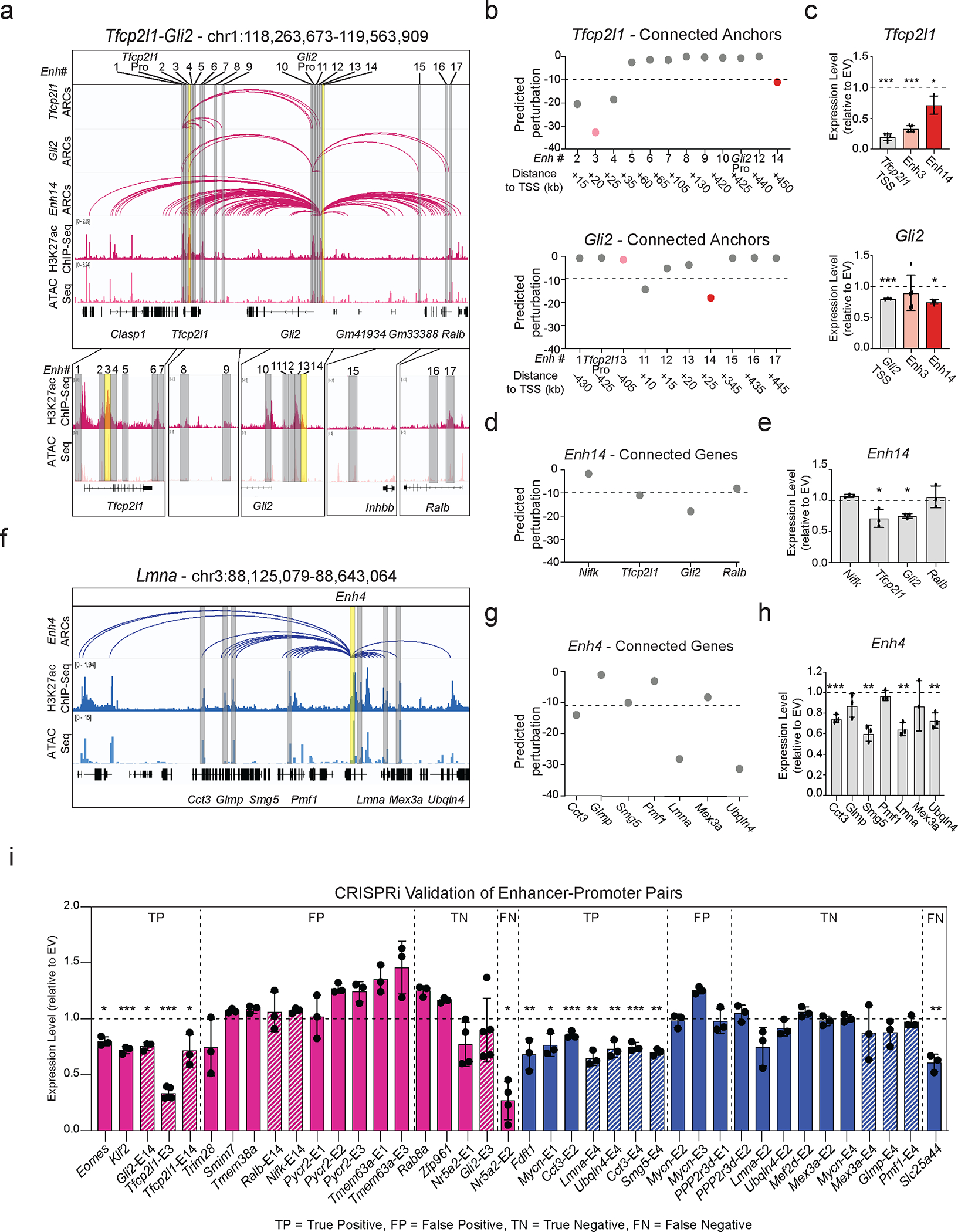
Mammalian embryogenesis commences with two pivotal and binary cell fate decisions that give rise to three essential lineages: the trophectoderm, the epiblast and the primitive endoderm. Although key signaling pathways and transcription factors that control these early embryonic decisions have been identified, the non-coding regulatory elements through which transcriptional regulators enact these fates remain understudied. Here, we characterize, at a genome-wide scale, enhancer activity and 3D connectivity in embryo-derived stem cell lines that represent each of the early developmental fates. We observe extensive enhancer remodeling and fine-scale 3D chromatin rewiring among the three lineages, which strongly associate with transcriptional changes, although distinct groups of genes are irresponsive to topological changes. In each lineage, a high degree of connectivity, or 'hubness', positively correlates with levels of gene expression and enriches for cell-type specific and essential genes. Genes within 3D hubs also show a significantly stronger probability of coregulation across lineages compared to genes in linear proximity or within the same contact domains. By incorporating 3D chromatin features, we build a predictive model for transcriptional regulation (3D-HiChAT) that outperforms models using only 1D promoter or proximal variables to predict levels and cell-type specificity of gene expression. Using 3D-HiChAT, we identify, in silico, candidate functional enhancers and hubs in each cell lineage, and with CRISPRi experiments, we validate several enhancers that control gene expression in their respective lineages. Our study identifies 3D regulatory hubs associated with the earliest mammalian lineages and describes their relationship to gene expression and cell identity, providing a framework to comprehensively understand lineage-specific transcriptional behaviors.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Conflict of interest statement
The authors declare that the above study was conducted in the absence of any commercial, financial, or personal relationships that could have appeared to influence the work reported in this article. All authors have approved the submitted version.
Figures
Update of
-
Systematic mapping and modeling of 3D enhancer-promoter interactions in early mouse embryonic lineages reveal regulatory principles that determine the levels and cell-type specificity of gene expression.bioRxiv [Preprint]. 2023 Jul 19:2023.07.19.549714. doi: 10.1101/2023.07.19.549714. bioRxiv. 2023. Update in: Nat Struct Mol Biol. 2024 Jan;31(1):125-140. doi: 10.1038/s41594-023-01130-4. PMID: 37577543 Free PMC article. Updated. Preprint.
Similar articles
-
Systematic mapping and modeling of 3D enhancer-promoter interactions in early mouse embryonic lineages reveal regulatory principles that determine the levels and cell-type specificity of gene expression.bioRxiv [Preprint]. 2023 Jul 19:2023.07.19.549714. doi: 10.1101/2023.07.19.549714. bioRxiv. 2023. Update in: Nat Struct Mol Biol. 2024 Jan;31(1):125-140. doi: 10.1038/s41594-023-01130-4. PMID: 37577543 Free PMC article. Updated. Preprint.
-
Chromatin connectivity maps reveal dynamic promoter-enhancer long-range associations.Nature. 2013 Dec 12;504(7479):306-310. doi: 10.1038/nature12716. Epub 2013 Nov 10. Nature. 2013. PMID: 24213634 Free PMC article.
-
Functional characterisation of cis-regulatory elements governing dynamic Eomes expression in the early mouse embryo.Development. 2017 Apr 1;144(7):1249-1260. doi: 10.1242/dev.147322. Epub 2017 Feb 7. Development. 2017. PMID: 28174238 Free PMC article.
-
Regulatory Landscaping: How Enhancer-Promoter Communication Is Sculpted in 3D.Mol Cell. 2019 Jun 20;74(6):1110-1122. doi: 10.1016/j.molcel.2019.05.032. Mol Cell. 2019. PMID: 31226276 Review.
-
3D organization of enhancers in MuSCs.Curr Top Dev Biol. 2024;158:407-431. doi: 10.1016/bs.ctdb.2024.01.011. Epub 2024 Mar 8. Curr Top Dev Biol. 2024. PMID: 38670714 Review.
Cited by
-
Predictive Prioritization of Enhancers Associated with Pancreas Disease Risk.bioRxiv [Preprint]. 2024 Sep 13:2024.09.07.611794. doi: 10.1101/2024.09.07.611794. bioRxiv. 2024. PMID: 39314336 Free PMC article. Preprint.
-
Synergistic and antagonistic activities of IRF8 and FOS enhancer pairs during an immune-cell fate switch.EMBO J. 2025 Apr;44(7):2025-2055. doi: 10.1038/s44318-025-00380-w. Epub 2025 Feb 19. EMBO J. 2025. PMID: 39972074 Free PMC article.
-
Single-cell analysis of bidirectional reprogramming between early embryonic states identify mechanisms of differential lineage plasticities in mice.Dev Cell. 2025 Mar 24;60(6):901-917.e12. doi: 10.1016/j.devcel.2024.11.022. Epub 2024 Dec 26. Dev Cell. 2025. PMID: 39729987 Free PMC article.
-
Three-dimensional regulatory hubs support oncogenic programs in glioblastoma.Mol Cell. 2025 Apr 3;85(7):1330-1348.e6. doi: 10.1016/j.molcel.2025.03.007. Epub 2025 Mar 26. Mol Cell. 2025. PMID: 40147440
-
A 3D genome view of colon cancer initiation.Nat Cancer. 2024 Nov;5(11):1600-1601. doi: 10.1038/s43018-024-00825-x. Nat Cancer. 2024. PMID: 39478116 No abstract available.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
