

Optimization of base editors for the functional correction of SMN2 as a treatment for spinal muscular atrophy
- PMID: 38057426
- PMCID: PMC10922509
- DOI: 10.1038/s41551-023-01132-z
Optimization of base editors for the functional correction of SMN2 as a treatment for spinal muscular atrophy
Abstract
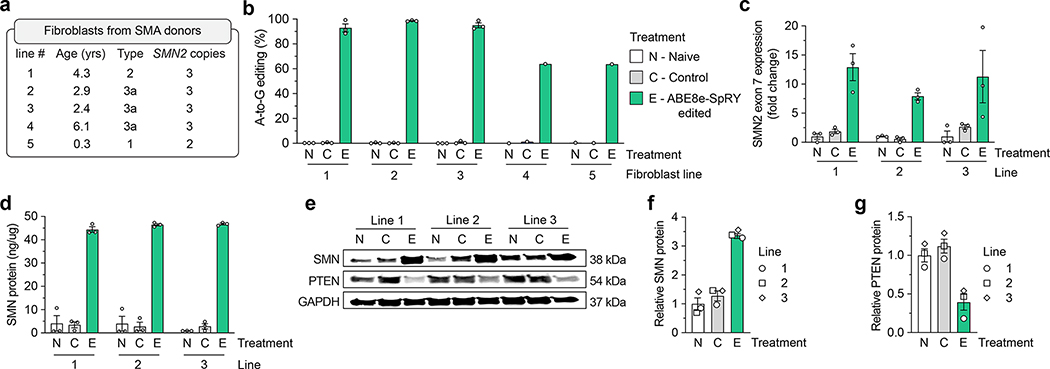
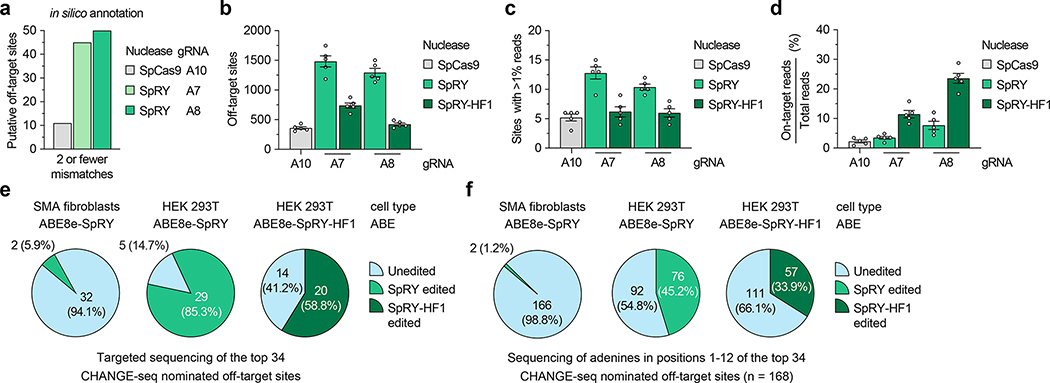
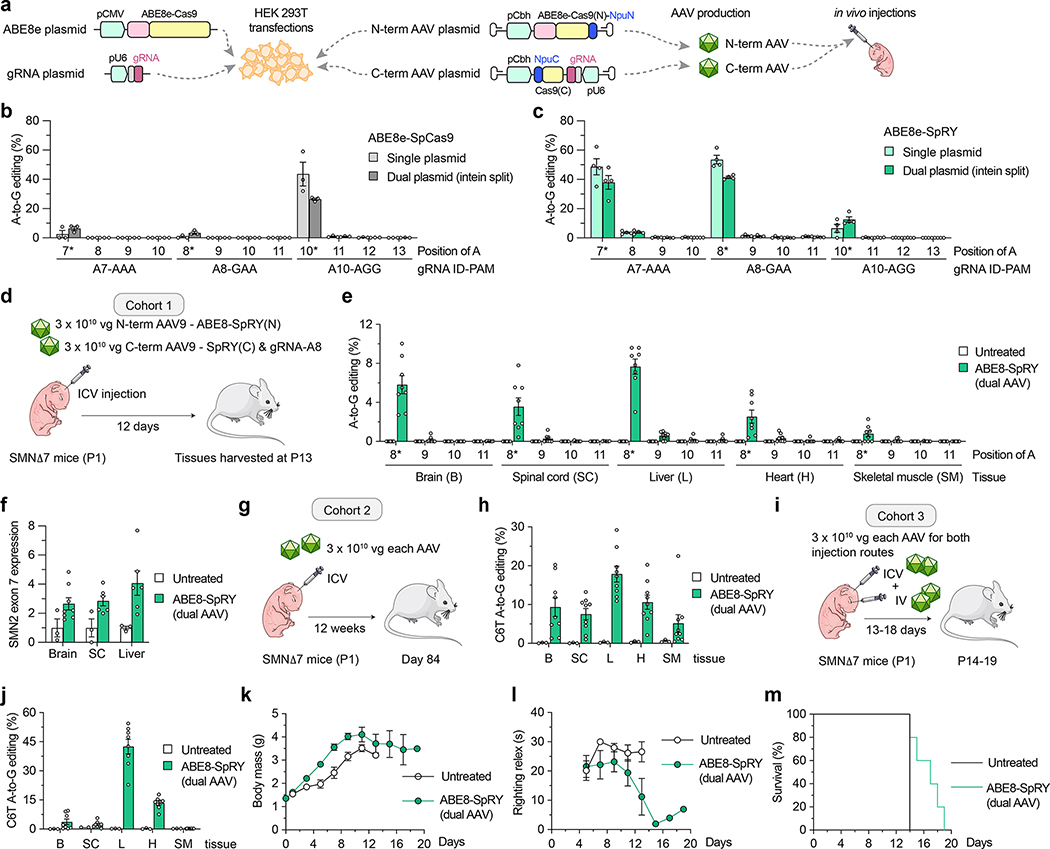
Spinal muscular atrophy (SMA) is caused by mutations in SMN1. SMN2 is a paralogous gene with a C•G-to-T•A transition in exon 7, which causes this exon to be skipped in most SMN2 transcripts, and results in low levels of the protein survival motor neuron (SMN). Here we show, in fibroblasts derived from patients with SMA and in a mouse model of SMA that, irrespective of the mutations in SMN1, adenosine base editors can be optimized to target the SMN2 exon-7 mutation or nearby regulatory elements to restore the normal expression of SMN. After optimizing and testing more than 100 guide RNAs and base editors, and leveraging Cas9 variants with high editing fidelity that are tolerant of different protospacer-adjacent motifs, we achieved the reversion of the exon-7 mutation via an A•T-to-G•C edit in up to 99% of fibroblasts, with concomitant increases in the levels of the SMN2 exon-7 transcript and of SMN. Targeting the SMN2 exon-7 mutation via base editing or other CRISPR-based methods may provide long-lasting outcomes to patients with SMA.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing interests
C.R.R.A., K.A.C., K.J.S., and B.P.K. are inventors on a patent application filed by Mass General Brigham (MGB) that describes genome engineering technologies to treat SMA. S.Q.T. and C.R.L are co-inventors on a patent application describing the CHANGE-seq method. S.Q.T. is a member of the scientific advisory board of Kromatid, Twelve Bio, and Prime Medicine. C.A.M. has a financial interest in Sphere Gene Therapeutics, Inc., Chameleon Biosciences, Inc., and Skylark Bio, Inc., companies developing gene therapy platforms. C.A.M.’s interests were reviewed and are managed by MGH and MGB in accordance with their conflict-of-interest policies. C.A.M. has a filed patent application with claims involving the AAV-F capsid. B.P.K. is an inventor on additional patents or patent applications filed by MGB that describe genome engineering technologies. B.P.K. is a consultant for EcoR1 capital and is on the scientific advisory board of Acrigen Biosciences, Life Edit Therapeutics, and Prime Medicine. S.Q.T. and B.P.K. have financial interests in Prime Medicine, Inc., a company developing therapeutic CRISPR-Cas technologies for gene editing. B.P.K.’s interests were reviewed and are managed by MGH and MGB in accordance with their conflict-of-interest policies. The other authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
