

Inferring compound heterozygosity from large-scale exome sequencing data
- PMID: 38057443
- PMCID: PMC10872287
- DOI: 10.1038/s41588-023-01608-3
Inferring compound heterozygosity from large-scale exome sequencing data
Abstract
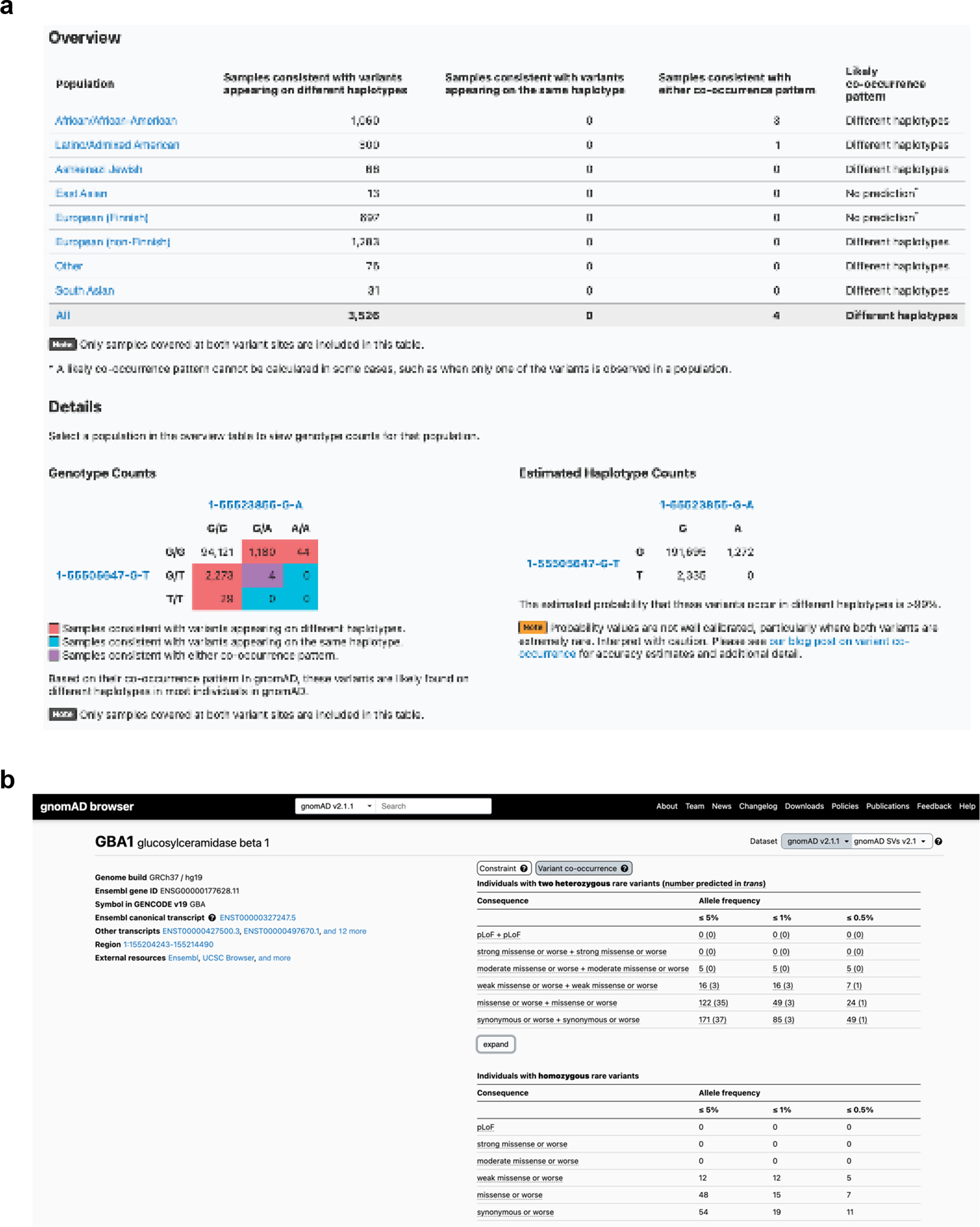
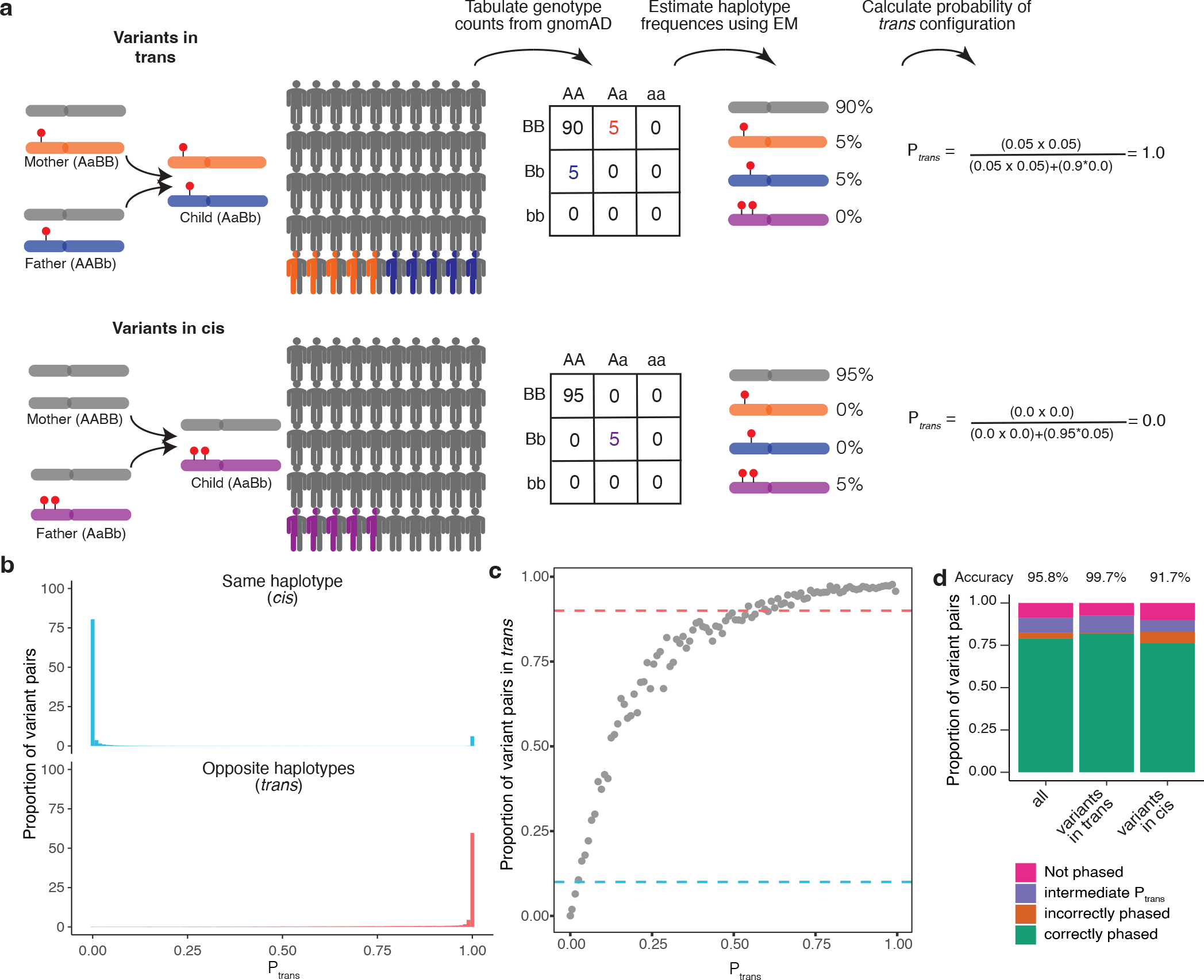
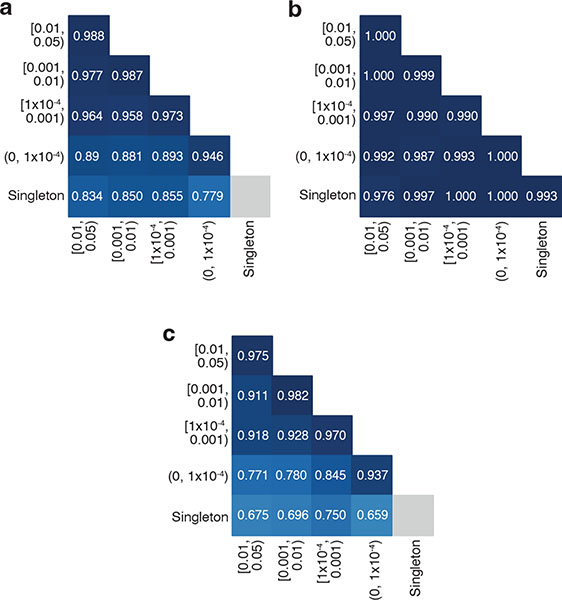
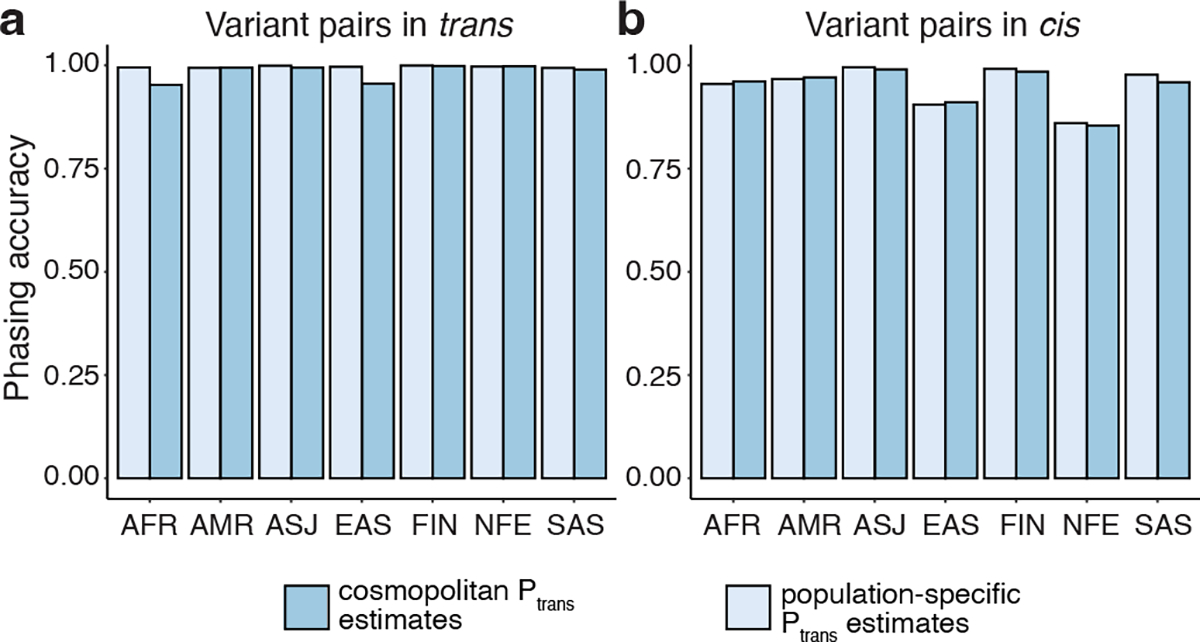
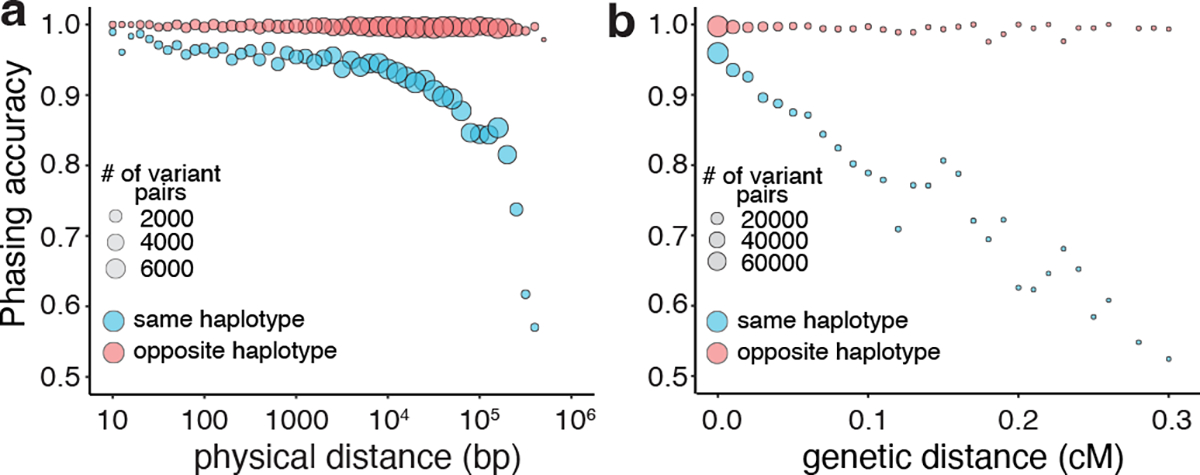
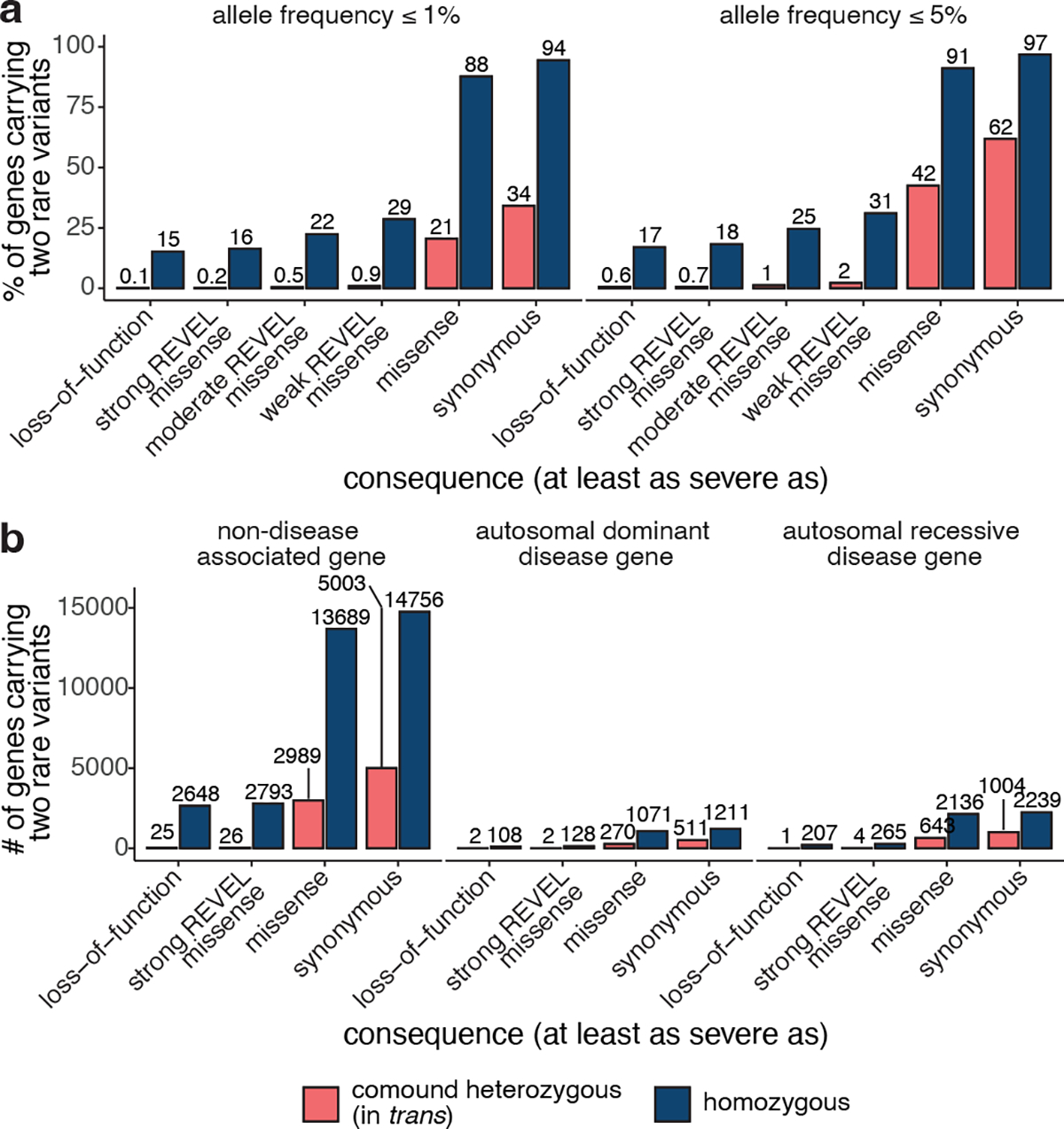
Recessive diseases arise when both copies of a gene are impacted by a damaging genetic variant. When a patient carries two potentially causal variants in a gene, accurate diagnosis requires determining that these variants occur on different copies of the chromosome (that is, are in trans) rather than on the same copy (that is, in cis). However, current approaches for determining phase, beyond parental testing, are limited in clinical settings. Here we developed a strategy for inferring phase for rare variant pairs within genes, leveraging genotypes observed in the Genome Aggregation Database (v2, n = 125,748 exomes). Our approach estimates phase with 96% accuracy, both in trio data and in patients with Mendelian conditions and presumed causal compound heterozygous variants. We provide a public resource of phasing estimates for coding variants and counts per gene of rare variants in trans that can aid interpretation of rare co-occurring variants in the context of recessive disease.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
Update of
-
Inferring compound heterozygosity from large-scale exome sequencing data.bioRxiv [Preprint]. 2023 Aug 21:2023.03.19.533370. doi: 10.1101/2023.03.19.533370. bioRxiv. 2023. Update in: Nat Genet. 2024 Jan;56(1):152-161. doi: 10.1038/s41588-023-01608-3. PMID: 36993580 Free PMC article. Updated. Preprint.
References
-
- Patterson M et al. WhatsHap: Weighted Haplotype Assembly for Future-Generation Sequencing Reads. J. Comput. Biol. 22, 498–509 (2015). - PubMed
Methods-only references
-
- Hail Team. Hail 0.2.105-acd89e80c345. GitHub; https://github.com/hail-is/hail/commit/acd89e80c345.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
