

Dictionary of immune responses to cytokines at single-cell resolution
- PMID: 38057668
- PMCID: PMC10781646
- DOI: 10.1038/s41586-023-06816-9
Dictionary of immune responses to cytokines at single-cell resolution
Abstract
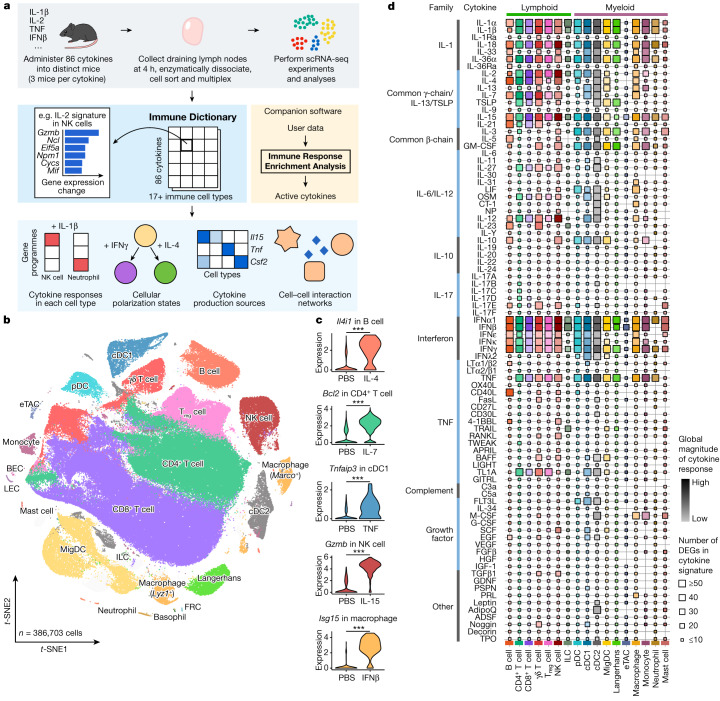
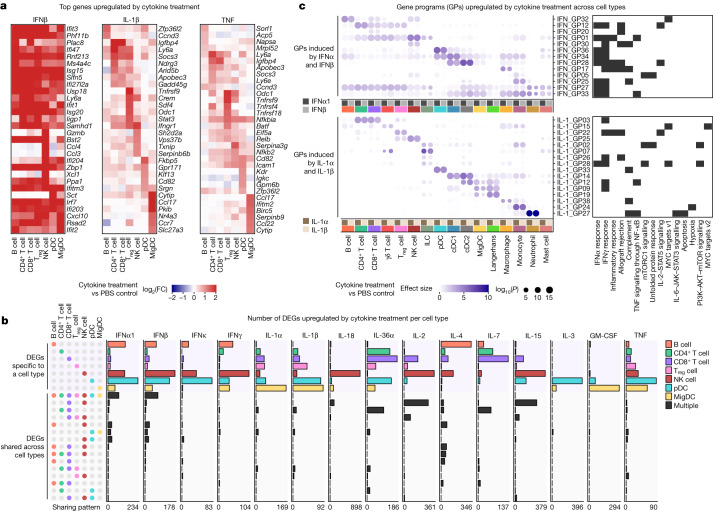
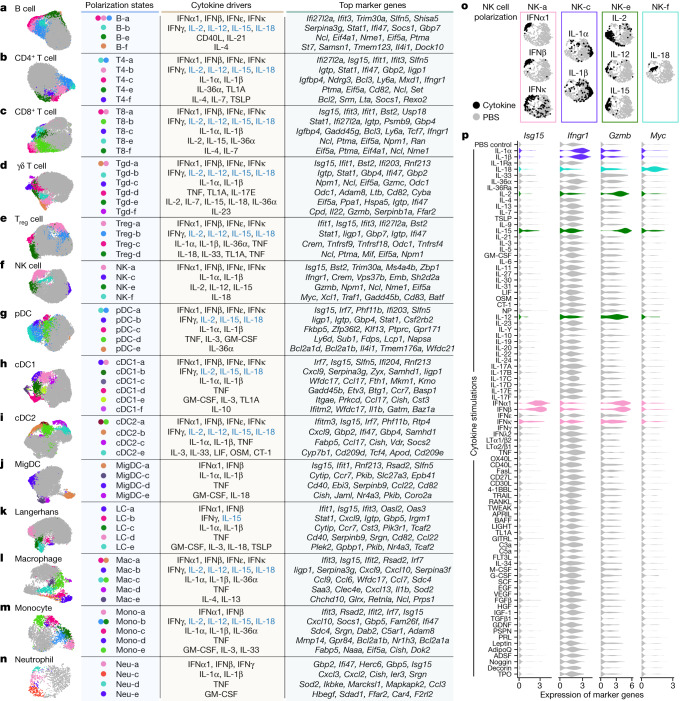
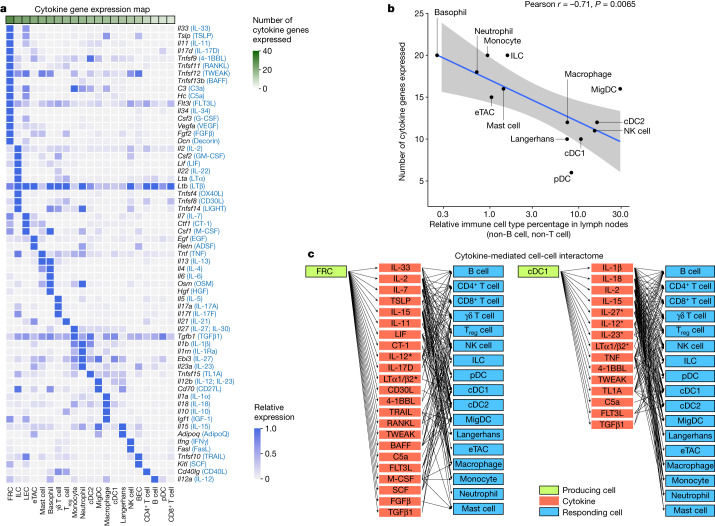
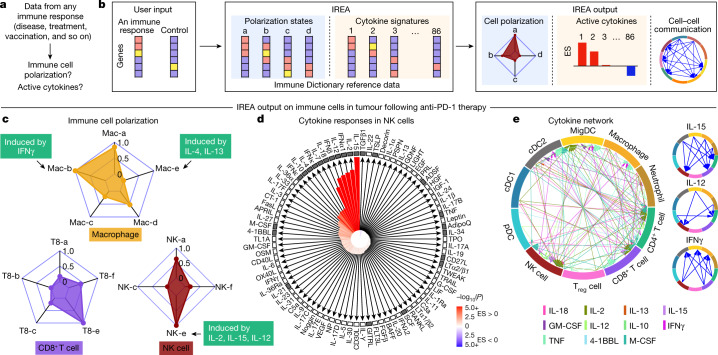
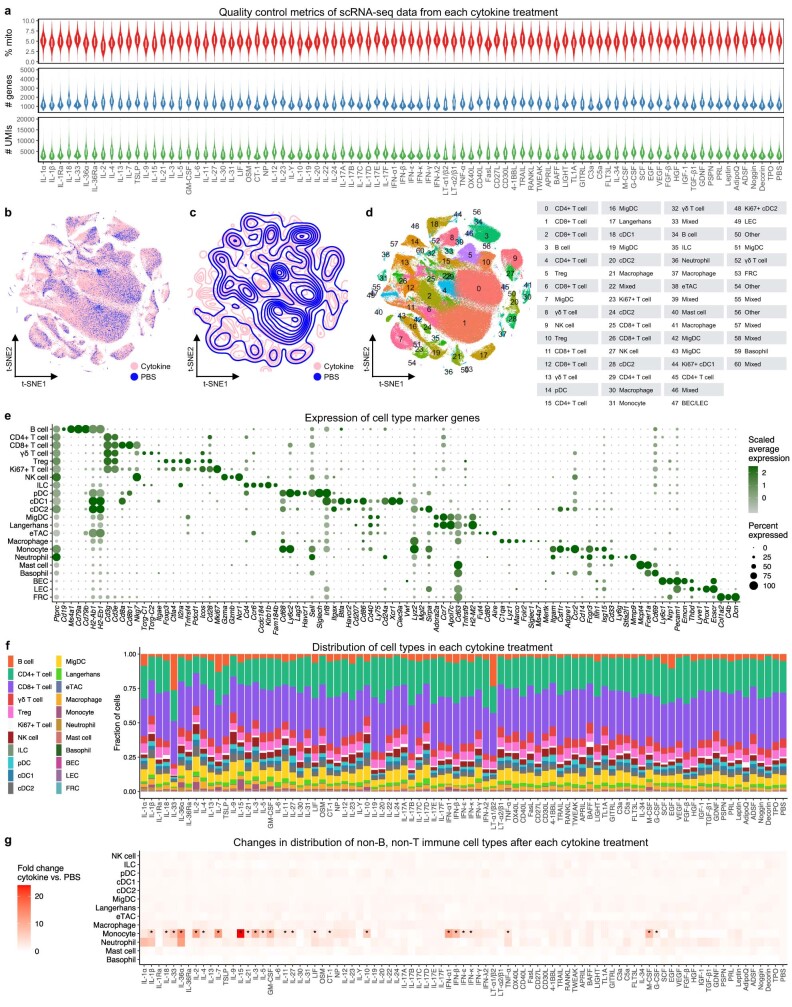
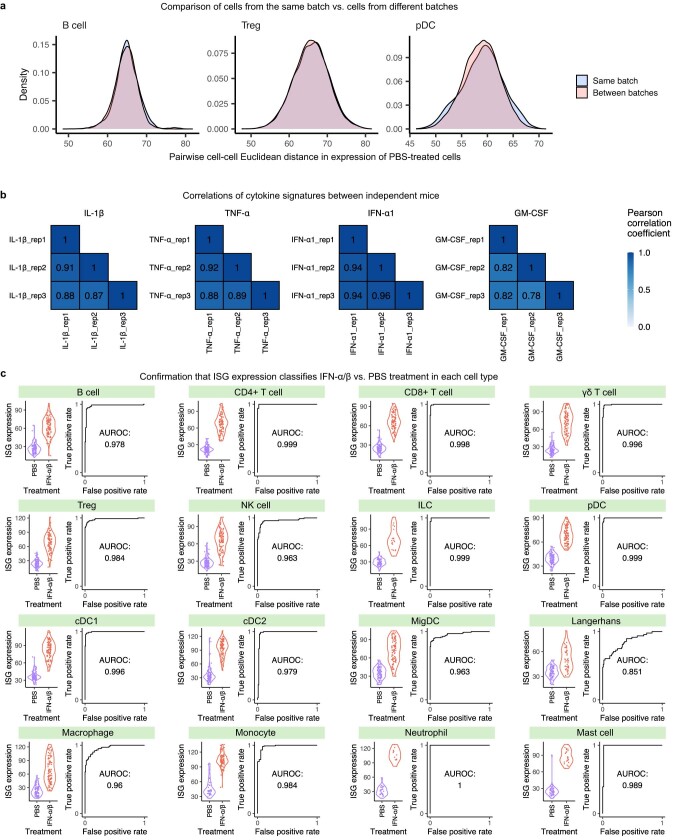
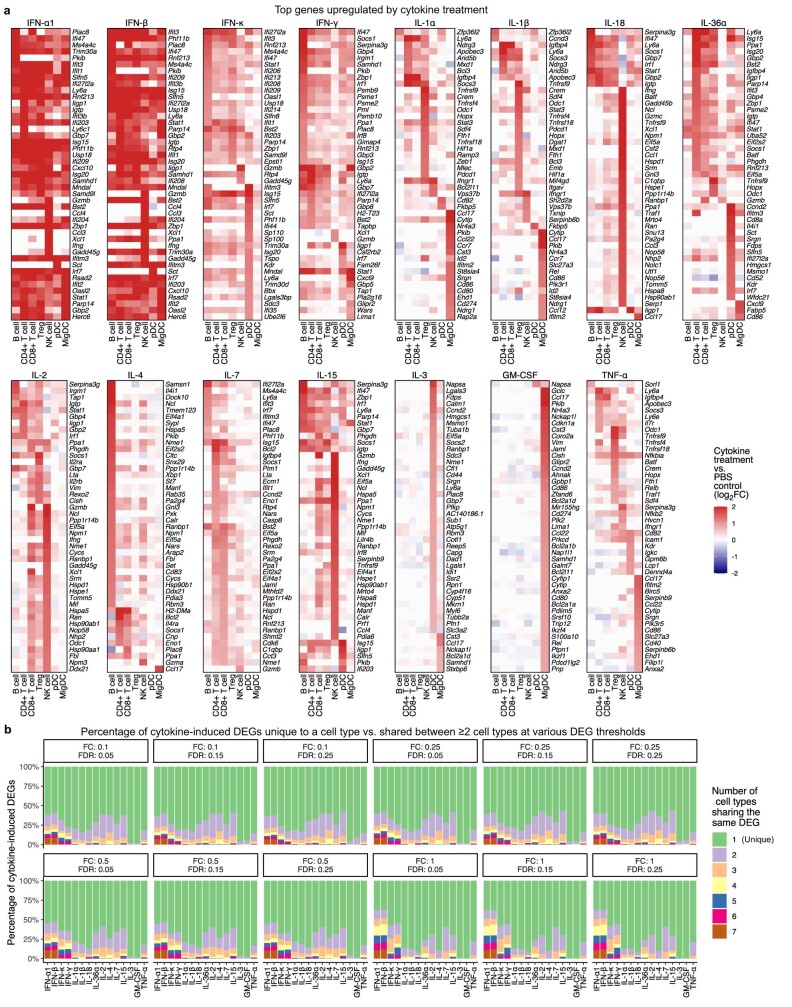
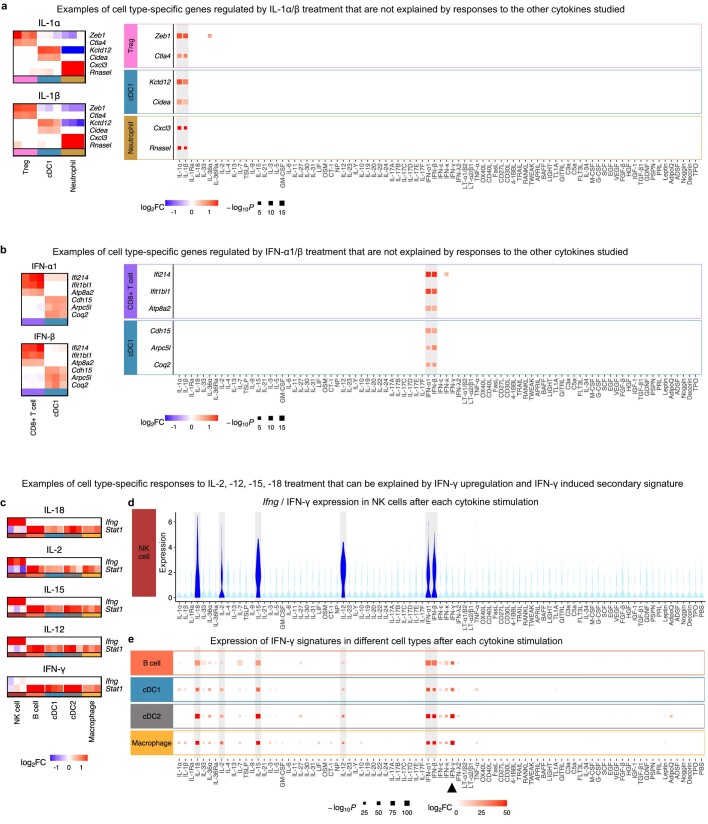
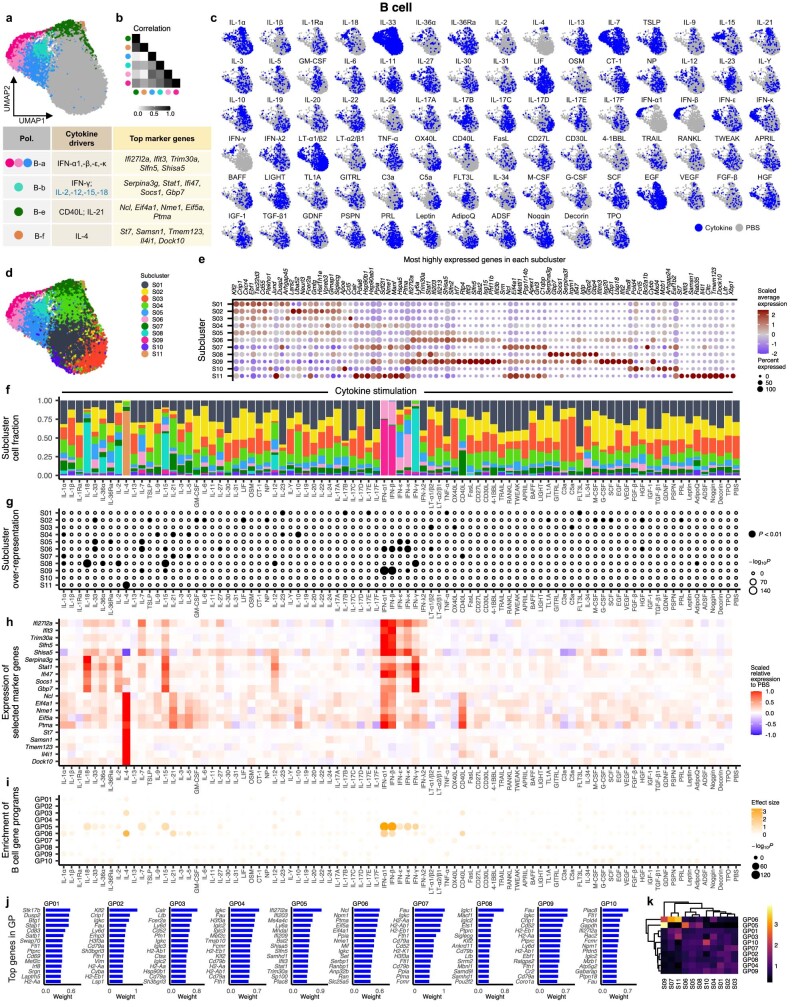
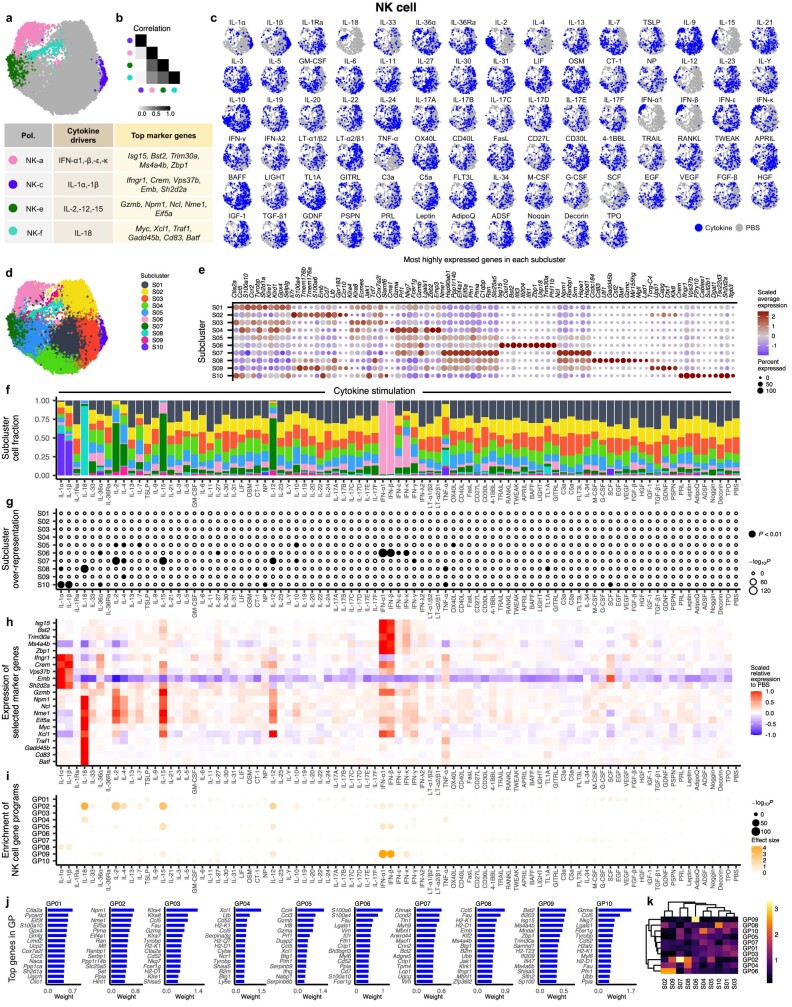
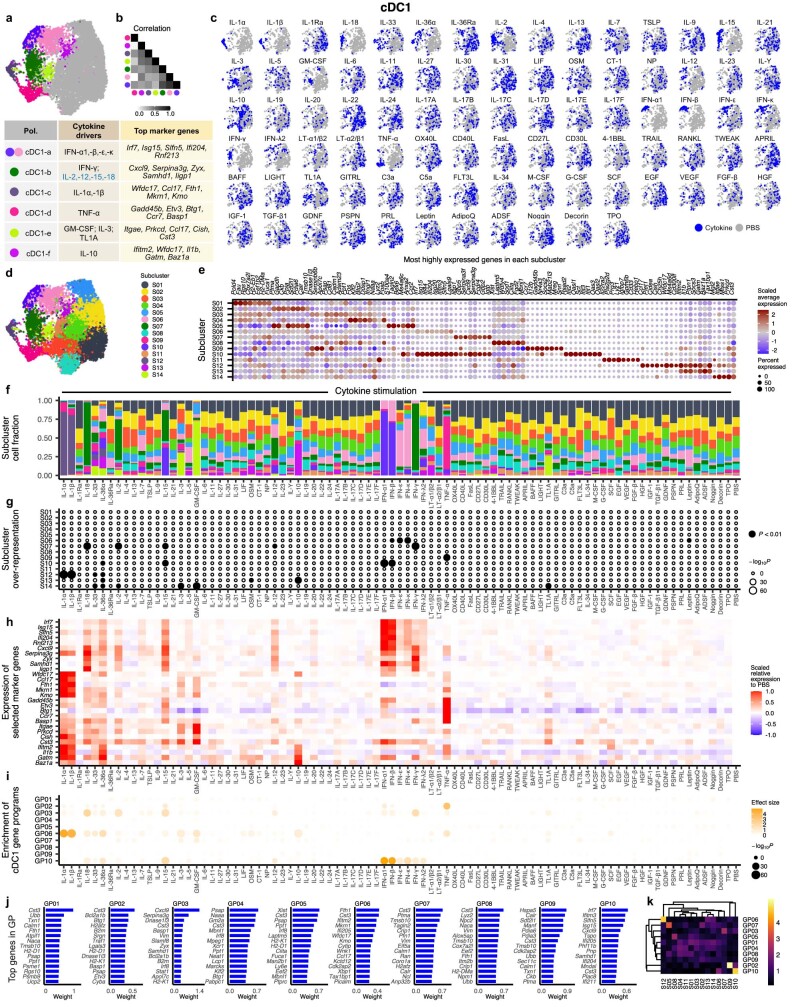
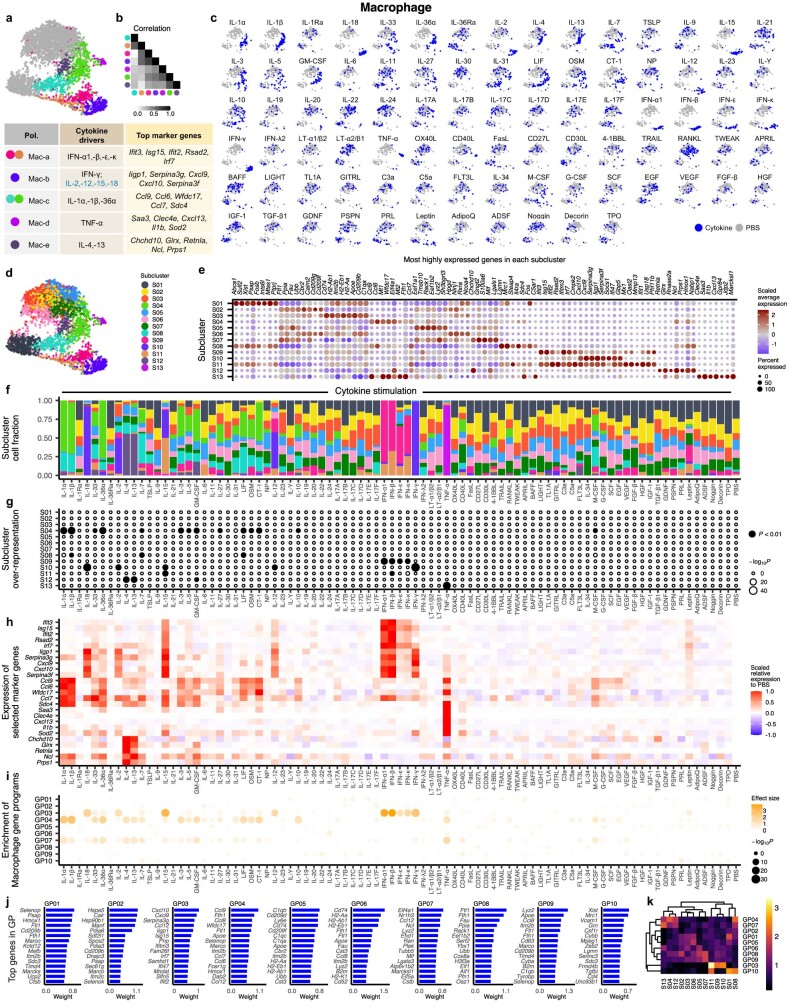
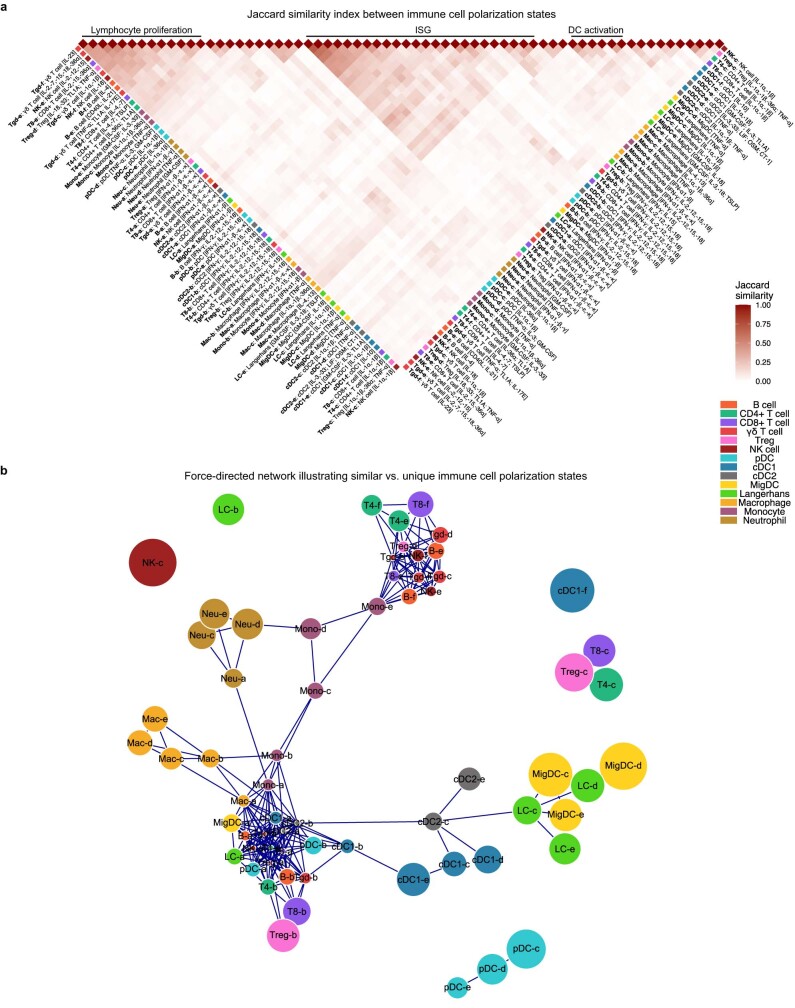
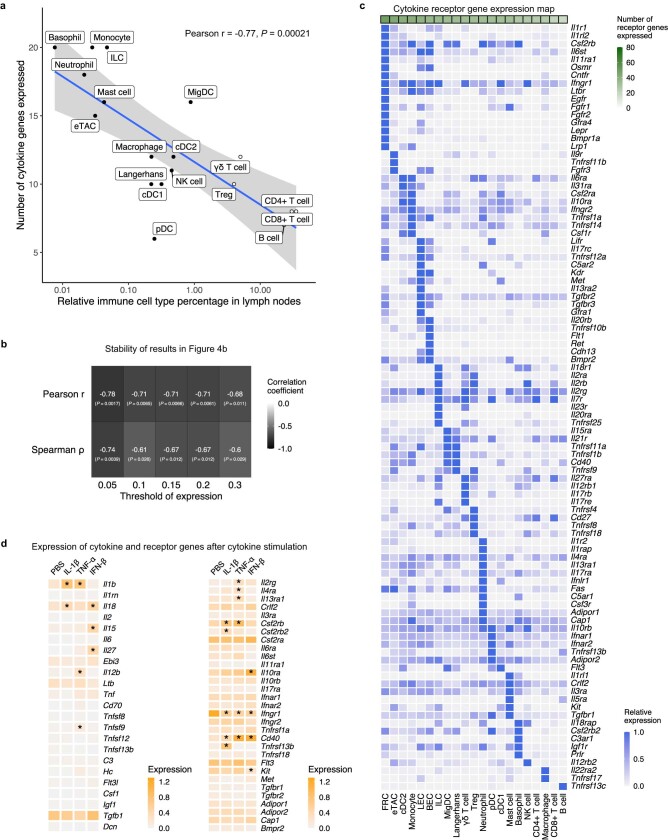
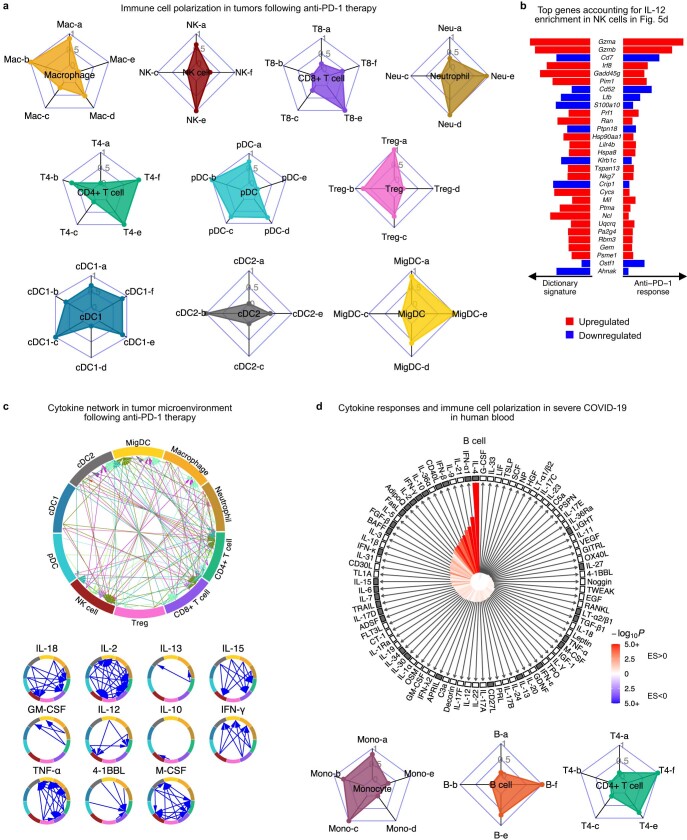
Cytokines mediate cell-cell communication in the immune system and represent important therapeutic targets1-3. A myriad of studies have highlighted their central role in immune function4-13, yet we lack a global view of the cellular responses of each immune cell type to each cytokine. To address this gap, we created the Immune Dictionary, a compendium of single-cell transcriptomic profiles of more than 17 immune cell types in response to each of 86 cytokines (>1,400 cytokine-cell type combinations) in mouse lymph nodes in vivo. A cytokine-centric view of the dictionary revealed that most cytokines induce highly cell-type-specific responses. For example, the inflammatory cytokine interleukin-1β induces distinct gene programmes in almost every cell type. A cell-type-centric view of the dictionary identified more than 66 cytokine-driven cellular polarization states across immune cell types, including previously uncharacterized states such as an interleukin-18-induced polyfunctional natural killer cell state. Based on this dictionary, we developed companion software, Immune Response Enrichment Analysis, for assessing cytokine activities and immune cell polarization from gene expression data, and applied it to reveal cytokine networks in tumours following immune checkpoint blockade therapy. Our dictionary generates new hypotheses for cytokine functions, illuminates pleiotropic effects of cytokines, expands our knowledge of activation states of each immune cell type, and provides a framework to deduce the roles of specific cytokines and cell-cell communication networks in any immune response.
© 2023. The Author(s).
Conflict of interest statement
N.H. and C.J.W. hold equity in BioNTech. N.H. is an advisor for Related Sciences/Danger Bio, Repertoire Immune Medicines and CytoReason. A.C. was a consultant for Foresite Capital and Altimmune for unrelated work. D.B.K is a scientific advisor for Immunitrack and Breakbio. DBK owns equity in Affimed N.V., Agenus, Armata Pharmaceuticals, Breakbio, BioMarin Pharmaceutical, Celldex Therapeutics, Editas Medicine, Gilead Sciences, Immunitybio and Lexicon Pharmaceuticals. BeiGene, a Chinese biotechnology company, supports unrelated research at the Translational Immunogenomics Lab. The remaining authors declare no competing interests. N.H. and A.C. have filed patent applications related to this work.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
