

∆4-3-oxo-5β-reductase deficiency: favorable outcome in 16 patients treated with cholic acid
- PMID: 38062451
- PMCID: PMC10704681
- DOI: 10.1186/s13023-023-02984-z
∆4-3-oxo-5β-reductase deficiency: favorable outcome in 16 patients treated with cholic acid
Abstract
Background: Oral cholic acid therapy is an effective therapy in children with primary bile acid synthesis deficiencies. Most reported patients with this treatment have 3β-hydroxy-Δ5-C27-steroid oxidoreductase deficiency. The aim of the study was the evaluation of cholic acid therapy in a cohort of patients with the rarer Δ4-3-oxosteroid 5β-reductase (Δ4-3-oxo-R) deficiency.
Methods: Sixteen patients with Δ4-3-oxo-R deficiency confirmed by AKR1D1 gene sequencing who received oral cholic acid were retrospectively analyzed.
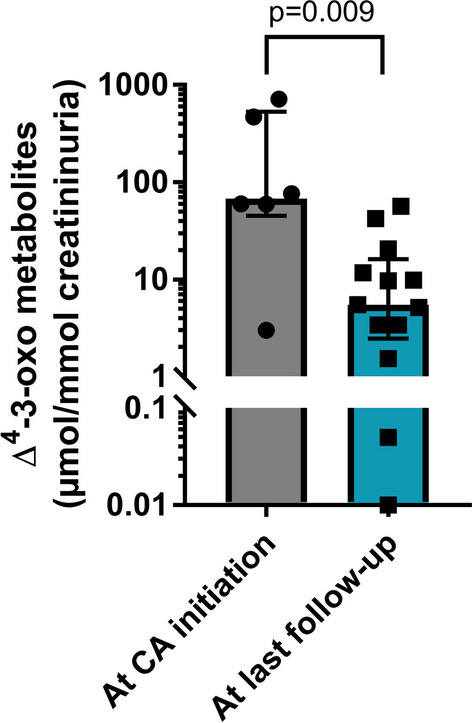
Results: First symptoms were reported early in life (median 2 months of age), with 14 and 3 patients having cholestatic jaundice and severe bleeding respectively. Fifteen patients received ursodeoxycholic acid before diagnosis, with partial improvement in 8 patients. Four patients had liver failure at the time of cholic acid initiation. All 16 patients received cholic acid from a median age of 8.1 months (range 3.1-159) and serum liver tests normalized in all within 6-12 months of treatment. After a median cholic acid therapy of 4.5 years (range 1.1-24), all patients were alive with their native liver. Median daily cholic acid dose at last follow-up was 8.3 mg/kg of body weight. All patients, but one, had normal physical examination and all had normal serum liver tests. Fibrosis, evaluated using liver biopsy (n = 4) or liver elastography (n = 9), had stabilized or improved. Cholic acid therapy enabled a 12-fold decrease of 3-oxo-∆4 derivatives in urine. Patients had normal growth and quality of life. The treatment was well tolerated without serious adverse events and signs of hepatotoxicity.
Conclusions: Oral cholic acid therapy is a safe and effective treatment for patients with Δ4-3-oxo-R deficiency.
Keywords: AKR1D1; Bile acid; Genetic cholestasis.
© 2023. The Author(s).
Conflict of interest statement
EJ is consultant for Vivet Therapeutics and Laboratoire CTRS. EG is consultant for Vivet Therapeutics, Laboratoire CTRS, Albireo, Mirum.
Figures
References
-
- Setchell KDR. Disorders of bile acid synthesis and metabolism. In: Suchy FJ, Sokol RJ, Balistreri WF, editors. Liver disease in children. 4. Cambridge: Cambridge University Press; 2014. pp. 567–586.
-
- Clayton PT, Leonard JV, Lawson AM, Setchell KD, Andersson S, Egestad B, et al. Familial giant cell hepatitis associated with synthesis of 3 beta, 7 alpha-dihydroxy-and 3 beta,7 alpha, 12 alpha-trihydroxy-5-cholenoic acids. J Clin Invest. 1987;79:1031–1038. doi: 10.1172/JCI112915. - DOI - PMC - PubMed
-
- Cheng JB, Jacquemin E, Gerhardt M, Nazer H, Cresteil D, Heubi JE, et al. Molecular genetics of 3β-Hydroxy-Δ5-C27-steroid oxidoreductase deficiency in 16 patients with loss of bile acid synthesis and liver disease. J Clin Endocrinol Metab. 2003;88:1833–1841. doi: 10.1210/jc.2002-021580. - DOI - PubMed
-
- Setchell KD, Suchy FJ, Welsh MB, Zimmer-Nechemias L, Heubi J, Balistreri WF. Delta 4–3-oxosteroid 5 beta-reductase deficiency described in identical twins with neonatal hepatitis. A new inborn error in bile acid synthesis. J Clin Invest. 1988;82:2148–2157. doi: 10.1172/JCI113837. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
