

Imputation strategies for genomic prediction using nanopore sequencing
- PMID: 38066581
- PMCID: PMC10709982
- DOI: 10.1186/s12915-023-01782-0
Imputation strategies for genomic prediction using nanopore sequencing
Abstract
Background: Genomic prediction describes the use of SNP genotypes to predict complex traits and has been widely applied in humans and agricultural species. Genotyping-by-sequencing, a method which uses low-coverage sequence data paired with genotype imputation, is becoming an increasingly popular SNP genotyping method for genomic prediction. The development of Oxford Nanopore Technologies' (ONT) MinION sequencer has now made genotyping-by-sequencing portable and rapid. Here we evaluate the speed and accuracy of genomic predictions using low-coverage ONT sequence data in a population of cattle using four imputation approaches. We also investigate the effect of SNP reference panel size on imputation performance.
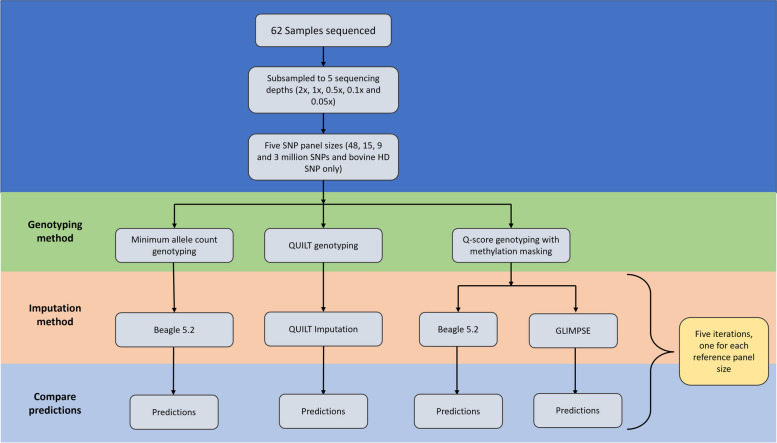
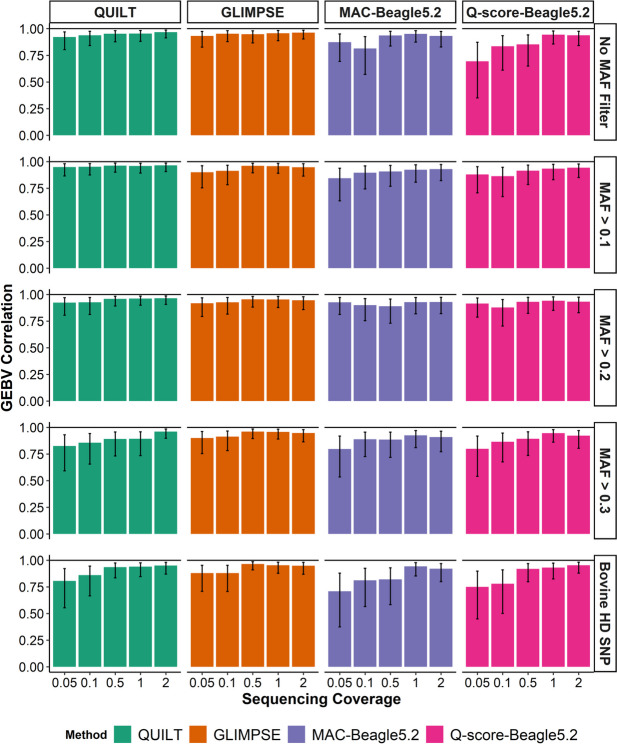
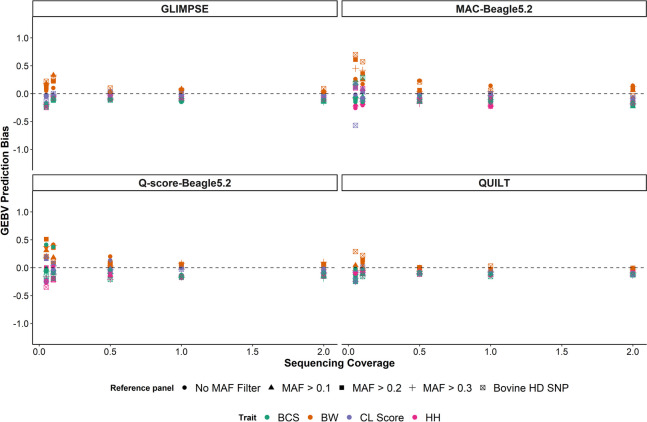
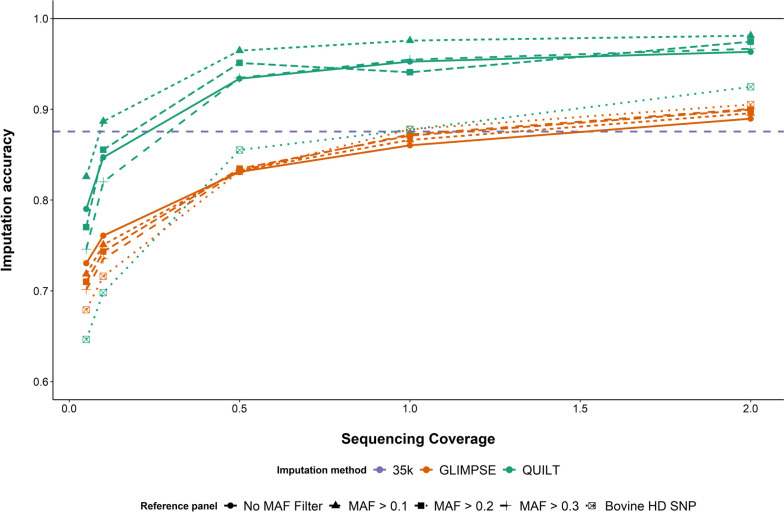
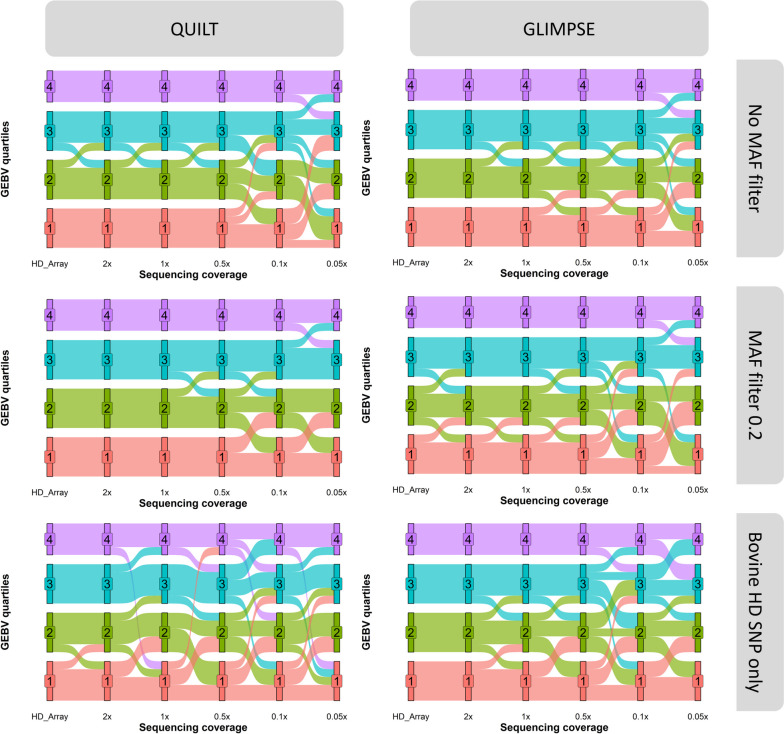
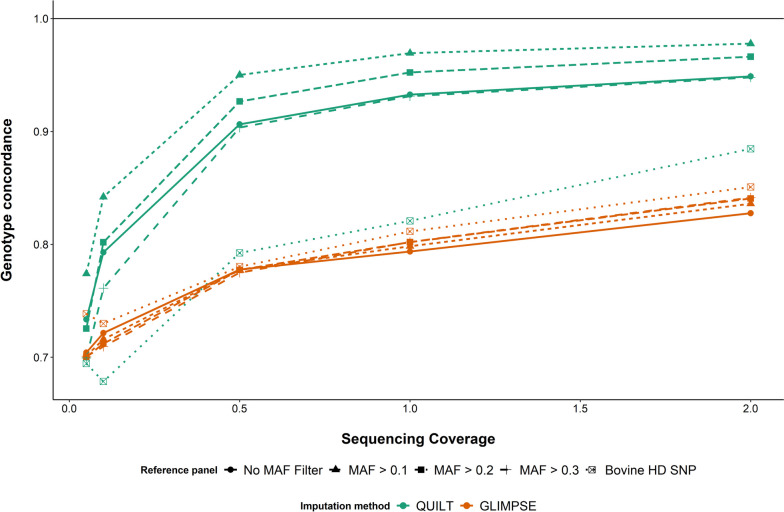
Results: SNP array genotypes and ONT sequence data for 62 beef heifers were used to calculate genomic estimated breeding values (GEBVs) from 641 k SNP for four traits. GEBV accuracy was much higher when genome-wide flanking SNP from sequence data were used to help impute the 641 k panel used for genomic predictions. Using the imputation package QUILT, correlations between ONT and low-density SNP array genomic breeding values were greater than 0.91 and up to 0.97 for sequencing coverages as low as 0.1 × using a reference panel of 48 million SNP. Imputation time was significantly reduced by decreasing the number of flanking sequence SNP used in imputation for all methods. When compared to high-density SNP arrays, genotyping accuracy and genomic breeding value correlations at 0.5 × coverage were also found to be higher than those imputed from low-density arrays.
Conclusions: Here we demonstrated accurate genomic prediction is possible with ONT sequence data from sequencing coverages as low as 0.1 × , and imputation time can be as short as 10 min per sample. We also demonstrate that in this population, genotyping-by-sequencing at 0.1 × coverage can be more accurate than imputation from low-density SNP arrays.
Keywords: Genomic prediction; Genotype imputation; Genotyping-by-sequencing; Oxford Nanopore Technologies sequencing; Skim-whole genome sequencing.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures



References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
