

New chromosome-scale genomes provide insights into marine adaptations of sea snakes (Hydrophis: Elapidae)
- PMID: 38066641
- PMCID: PMC10709897
- DOI: 10.1186/s12915-023-01772-2
New chromosome-scale genomes provide insights into marine adaptations of sea snakes (Hydrophis: Elapidae)
Abstract
Background: Sea snakes underwent a complete transition from land to sea within the last ~ 15 million years, yet they remain a conspicuous gap in molecular studies of marine adaptation in vertebrates.
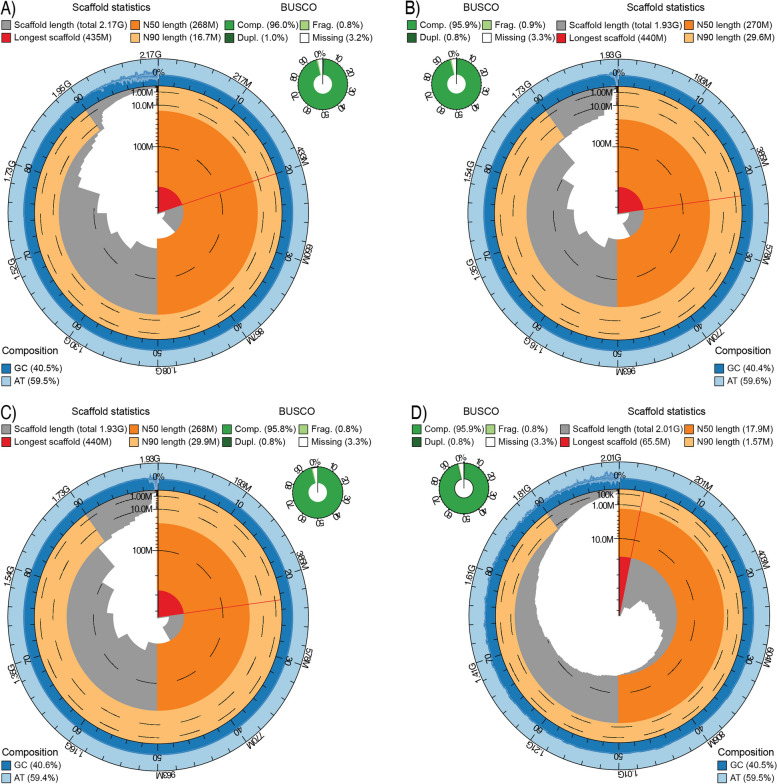
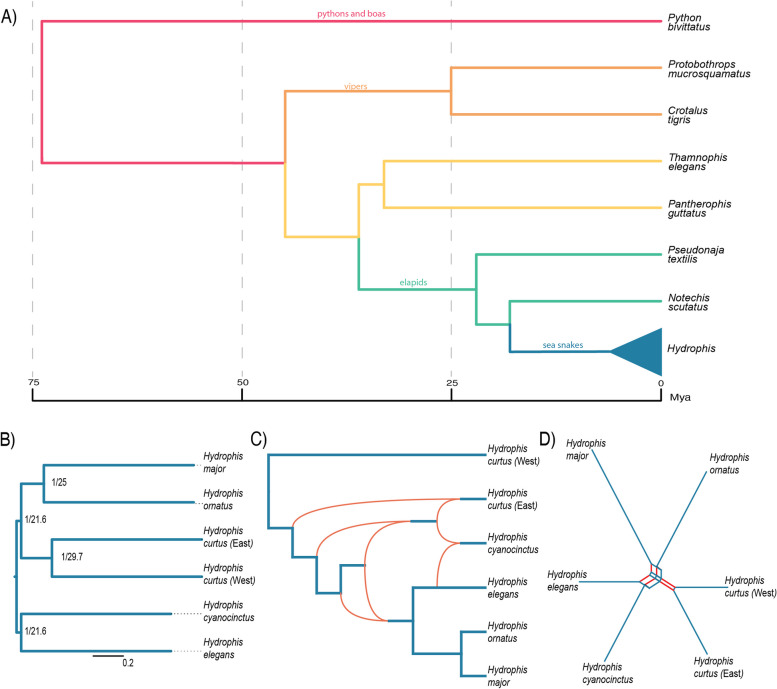
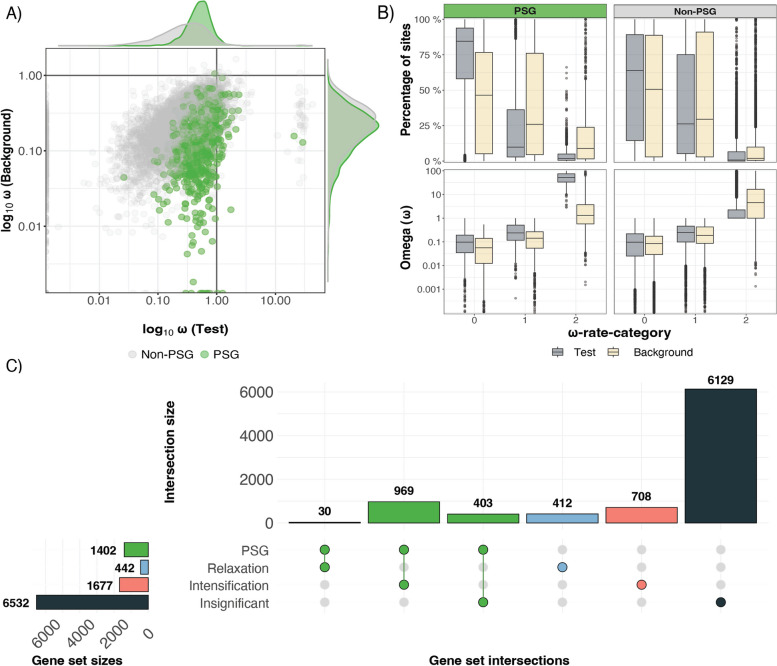
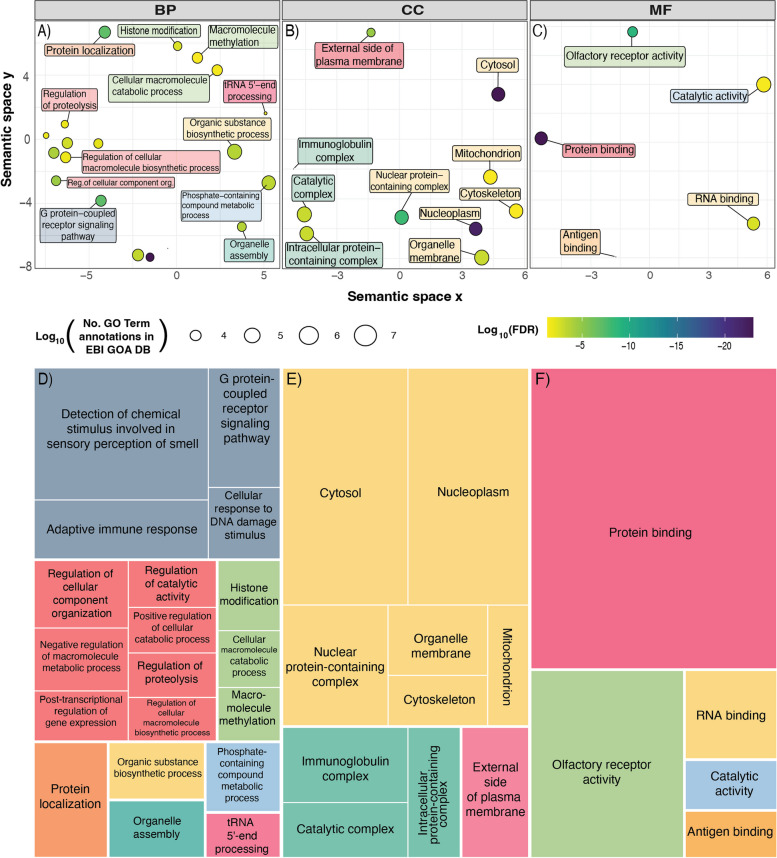
Results: Here, we generate four new annotated sea snake genomes, three of these at chromosome-scale (Hydrophis major, H. ornatus and H. curtus), and perform detailed comparative genomic analyses of sea snakes and their closest terrestrial relatives. Phylogenomic analyses highlight the possibility of near-simultaneous speciation at the root of Hydrophis, and synteny maps show intra-chromosomal variations that will be important targets for future adaptation and speciation genomic studies of this system. We then used a strict screen for positive selection in sea snakes (against a background of seven terrestrial snake genomes) to identify genes over-represented in hypoxia adaptation, sensory perception, immune response and morphological development.
Conclusions: We provide the best reference genomes currently available for the prolific and medically important elapid snake radiation. Our analyses highlight the phylogenetic complexity and conserved genome structure within Hydrophis. Positively selected marine-associated genes provide promising candidates for future, functional studies linking genetic signatures to the marine phenotypes of sea snakes and other vertebrates.
Keywords: Chromosome-scale genome; Marine adaptation; Positive selection; Sea snake; Synteny.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
