

Diagnostic Challenges in ABCA4-Associated Retinal Degeneration: One Gene, Many Phenotypes
- PMID: 38066771
- PMCID: PMC10706589
- DOI: 10.3390/diagnostics13233530
Diagnostic Challenges in ABCA4-Associated Retinal Degeneration: One Gene, Many Phenotypes
Abstract
(1) Purpose: ABCA4-associated retinal degeneration (ABCA4-RD) is a phenotypically diverse disease that often evades diagnosis, even by experienced retinal specialists. This may lead to inappropriate management, delayed genetic testing, or inaccurate interpretation of genetic testing results. Here, we illustrate the phenotypic diversity of ABCA4-RD using a series of representative cases and compare these to other conditions that closely mimic ABCA4-RD. (2) Methods: Genetically confirmed ABCA4-RD cases with representative phenotypes were selected from an inherited retinal disease cohort in Singapore and compared to phenocopies involving other retinal diseases. (3) Results: ABCA4-RD phenotypes in this series included typical adolescent-onset Stargardt disease with flecks, bull's eye maculopathy without flecks, fundus flavimaculatus, late-onset Stargardt disease, and severe early-onset Stargardt disease. Phenocopies of ABCA4-RD in this series included macular dystrophy, pattern dystrophy, cone dystrophy, advanced retinitis pigmentosa, Leber congenital amaurosis, drug toxicity, and age-related macular degeneration. Key distinguishing features that often suggested a diagnosis of ABCA4-RD were the presence of peripapillary sparing, macular involvement and centrifugal distribution, and a recessive pedigree. (4) Conclusions: ABCA4-RD demonstrates a remarkable phenotypic spectrum that makes diagnosis challenging. Awareness of the clinical spectrum of disease can facilitate prompt recognition and accurate diagnostic testing.
Keywords: ABCA4; Stargardt disease; genetic diagnosis; inherited retinal disease; phenotypic variation; retinal dystrophy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures




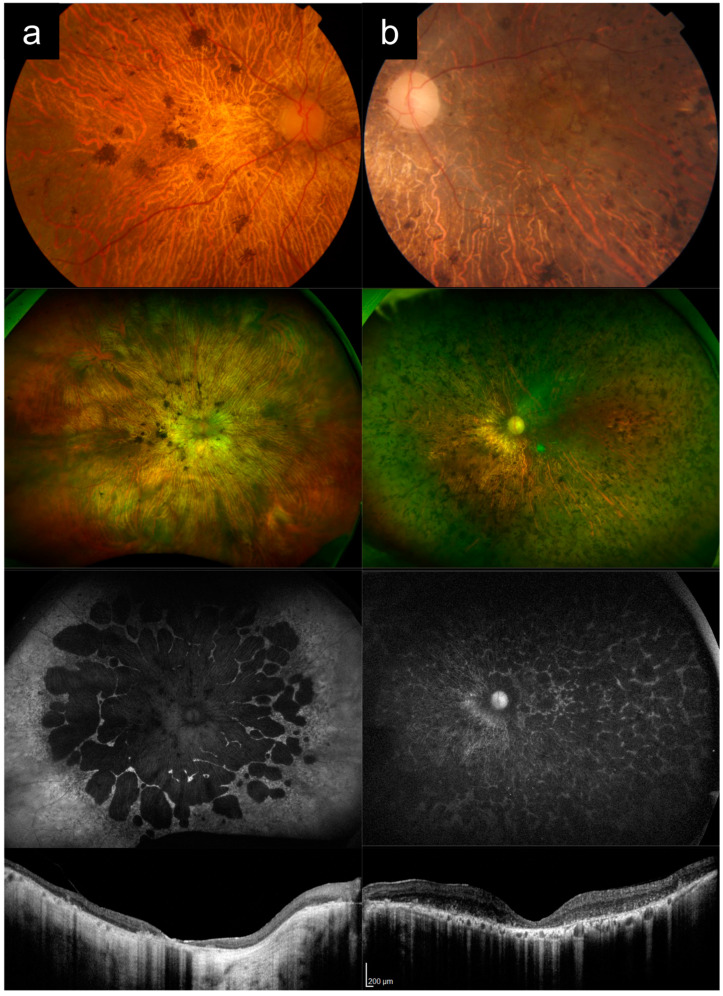
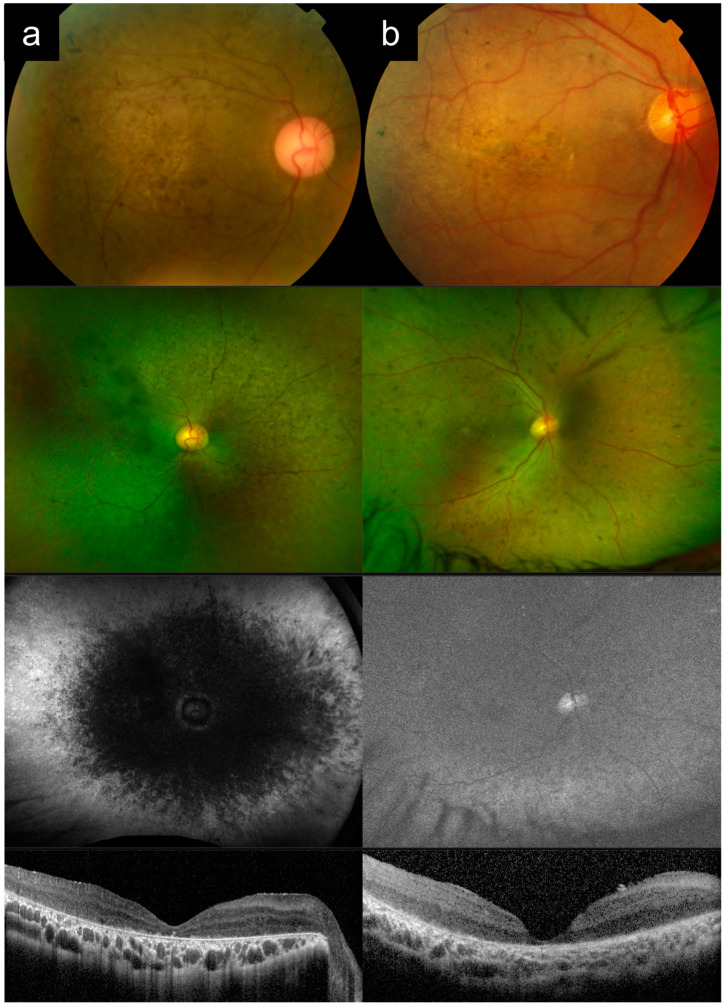
References
-
- Galvin O., Chi G., Brady L., Hippert C., Del Valle Rubido M., Daly A., Michaelides M. The Impact of Inherited Retinal Diseases in the Republic of Ireland (ROI) and the United Kingdom (UK) from a Cost-of-Illness Perspective. Clin. Ophthalmol. 2020;14:707–719. doi: 10.2147/OPTH.S241928. - DOI - PMC - PubMed
-
- Gong J., Cheung S., Fasso-Opie A., Galvin O., Moniz L.S., Earle D., Durham T., Menzo J., Li N., Duffy S., et al. The Impact of Inherited Retinal Diseases in the United States of America (US) and Canada from a Cost-of-Illness Perspective. Clin. Ophthalmol. 2021;15:2855–2866. doi: 10.2147/OPTH.S313719. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
