

Hotspots of Somatic Genetic Variation in Pituitary Neuroendocrine Tumors
- PMID: 38067388
- PMCID: PMC10705109
- DOI: 10.3390/cancers15235685
Hotspots of Somatic Genetic Variation in Pituitary Neuroendocrine Tumors
Abstract
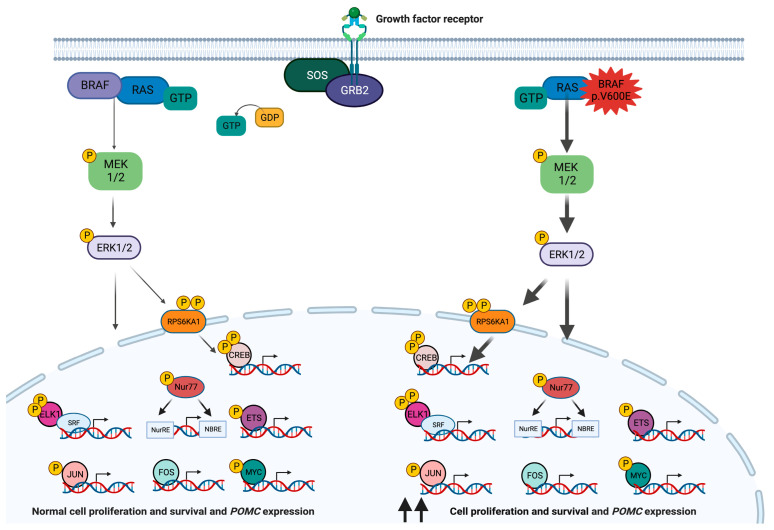
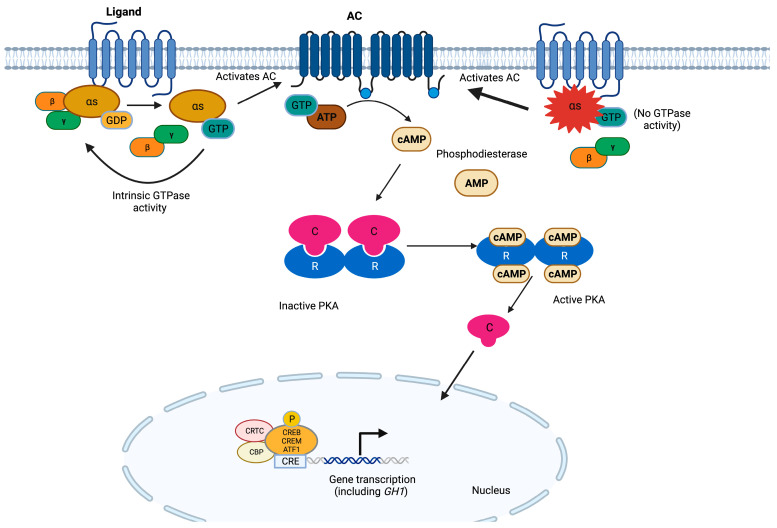
The most common genetic drivers of pituitary neuroendocrine tumors (PitNETs) lie within mutational hotspots, which are genomic regions where variants tend to cluster. Some of these hotspot defects are unique to PitNETs, while others are associated with additional neoplasms. Hotspot variants in GNAS and USP8 are the most common genetic causes of acromegaly and Cushing's disease, respectively. Although it has been proposed that these genetic defects could define specific clinical phenotypes, results are highly variable among studies. In contrast, DICER1 hotspot variants are associated with a familial syndrome of cancer predisposition, and only exceptionally occur as somatic changes. A small number of non-USP8-driven corticotropinomas are due to somatic hotspot variants in USP48 or BRAF; the latter is a well-known mutational hotspot in cancer. Finally, somatic variants affecting a hotspot in SF3B1 have been associated with multiple cancers and, more recently, with prolactinomas. Since the associations of BRAF, USP48, and SF3B1 hotspot variants with PitNETs are very recent, their effects on clinical phenotypes are still unknown. Further research is required to fully define the role of these genetic defects as disease biomarkers and therapeutic targets.
Keywords: druggable target; genetic driver; mutational hotspot; pituitary neuroendocrine tumor; somatic variant.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


Similar articles
-
Clinical Spectrum of USP8 Pathogenic Variants in Cushing's Disease.Arch Med Res. 2023 Dec;54(8):102899. doi: 10.1016/j.arcmed.2023.102899. Epub 2023 Nov 2. Arch Med Res. 2023. PMID: 37925320 Review.
-
Genetic Profiling of a Cohort of Italian Patients with ACTH-Secreting Pituitary Tumors and Characterization of a Novel USP8 Gene Variant.Cancers (Basel). 2021 Aug 10;13(16):4022. doi: 10.3390/cancers13164022. Cancers (Basel). 2021. PMID: 34439178 Free PMC article.
-
Driver mutations in USP8 wild-type Cushing's disease.Neuro Oncol. 2019 Oct 9;21(10):1273-1283. doi: 10.1093/neuonc/noz109. Neuro Oncol. 2019. PMID: 31222332 Free PMC article.
-
USP8, USP48, and BRAF mutations differ in their genotype-phenotype correlation in Asian Indian patients with Cushing's disease.Endocrine. 2022 Feb;75(2):549-559. doi: 10.1007/s12020-021-02903-x. Epub 2021 Oct 18. Endocrine. 2022. PMID: 34664215
-
Genomics and Epigenomics of Pituitary Tumors: What Do Pathologists Need to Know?Endocr Pathol. 2021 Mar;32(1):3-16. doi: 10.1007/s12022-021-09663-4. Epub 2021 Jan 12. Endocr Pathol. 2021. PMID: 33433883 Review.
Cited by
-
Advancements in Molecular Diagnosis and Pharmacotherapeutic Strategies for Invasive Pituitary Adenomas.Immun Inflamm Dis. 2024 Dec;12(12):e70098. doi: 10.1002/iid3.70098. Immun Inflamm Dis. 2024. PMID: 39688352 Free PMC article. Review.
-
A Study of Alternative TrkA Splicing Identifies TrkAIII as a Novel Potentially Targetable Participant in PitNET Progression.Biology (Basel). 2024 Mar 7;13(3):171. doi: 10.3390/biology13030171. Biology (Basel). 2024. PMID: 38534441 Free PMC article.
References
-
- Cooper D.N., Mort M., Stenson P.D., Ball E.V., Chuzhanova N.A. Methylation-mediated deamination of 5-methylcytosine appears to give rise to mutations causing human inherited disease in CpNpG trinucleotides, as well as in CpG dinucleotides. Hum. Genom. 2010;4:406–410. doi: 10.1186/1479-7364-4-6-406. - DOI - PMC - PubMed
Publication types
Grants and funding
- Departmental funds, Coordination of Scientific Research/Universidad Nacional Autónoma de México
- Support Program for Research Projects and Technological Innovation TA200322/Universidad Nacional Autónoma de México
- Financial support for research projects/Sociedad Mexicana de Nutrición y Endocrinología
- Equipment grant, 2022/Society for Endocrinology
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
