

Single cell multi-omics reveal intra-cell-line heterogeneity across human cancer cell lines
- PMID: 38071219
- PMCID: PMC10710513
- DOI: 10.1038/s41467-023-43991-9
Single cell multi-omics reveal intra-cell-line heterogeneity across human cancer cell lines
Abstract
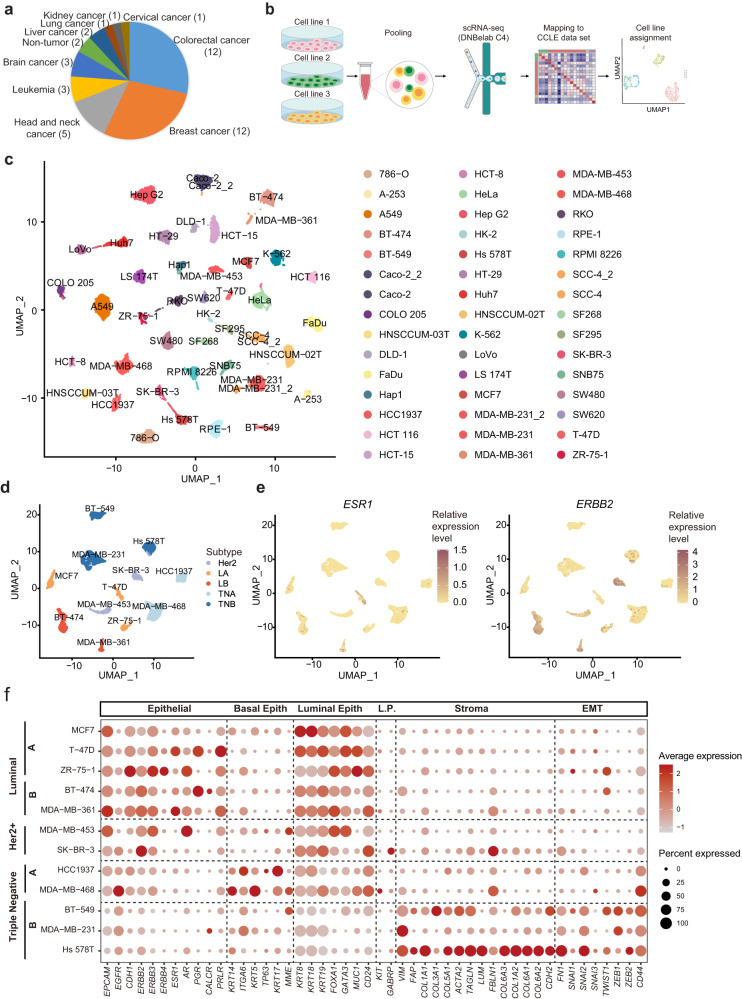
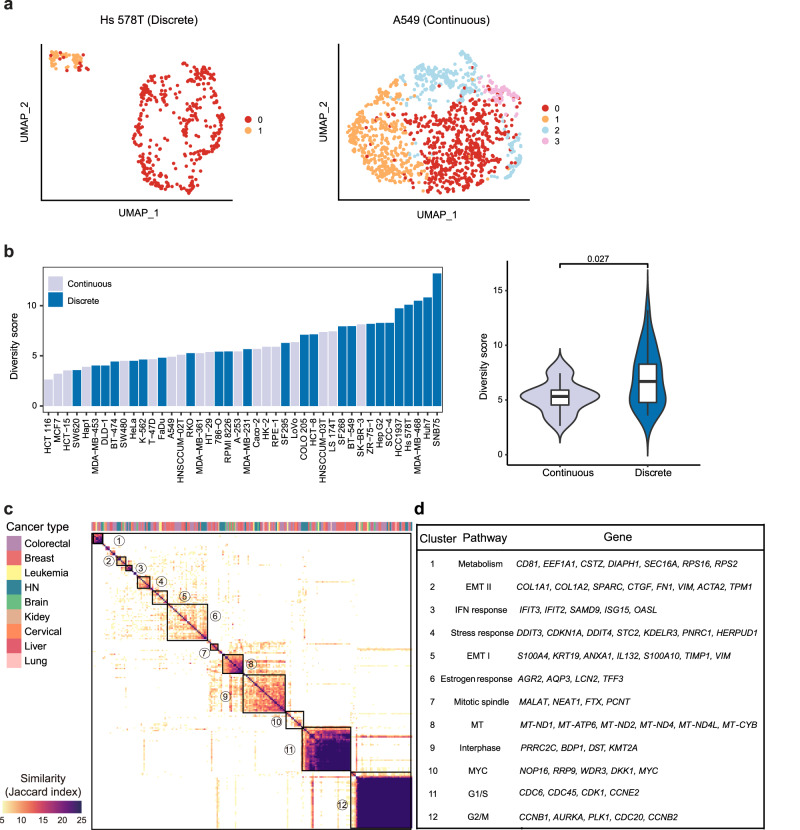
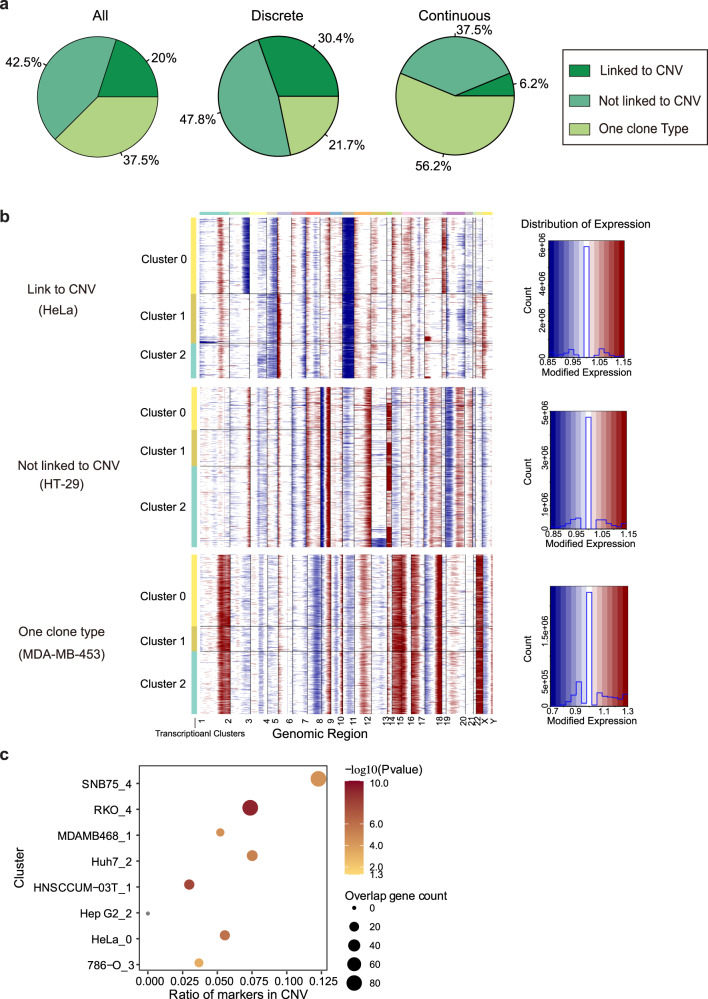
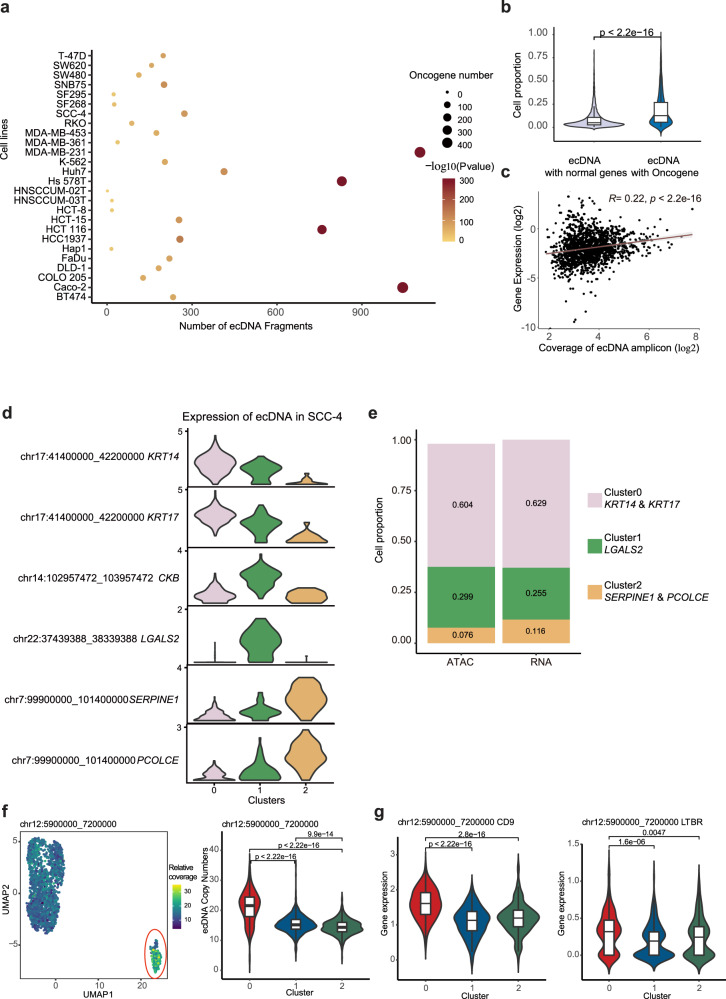
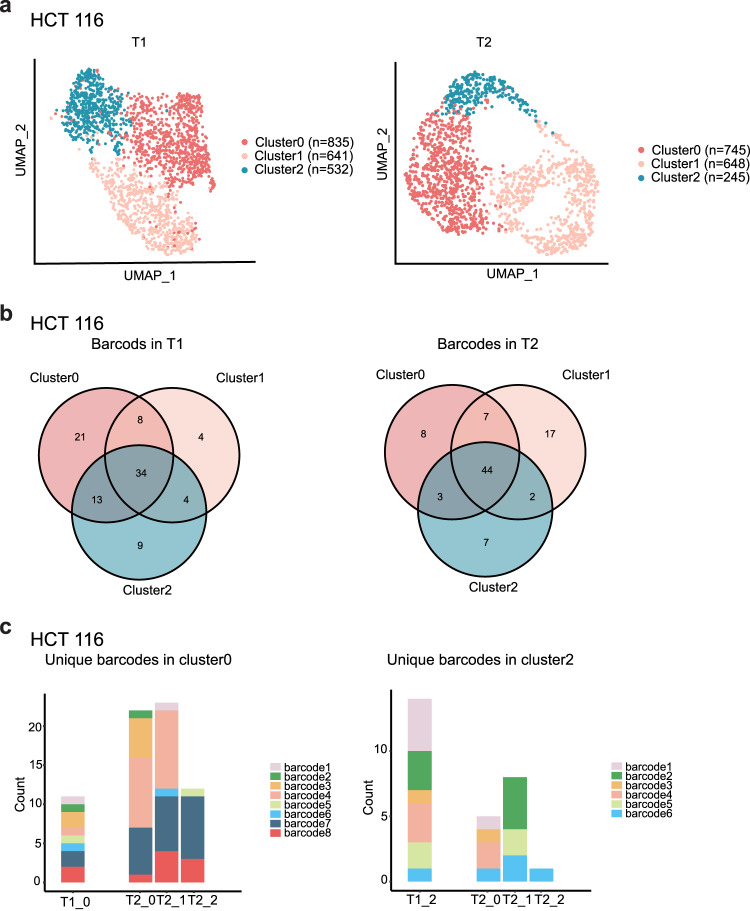
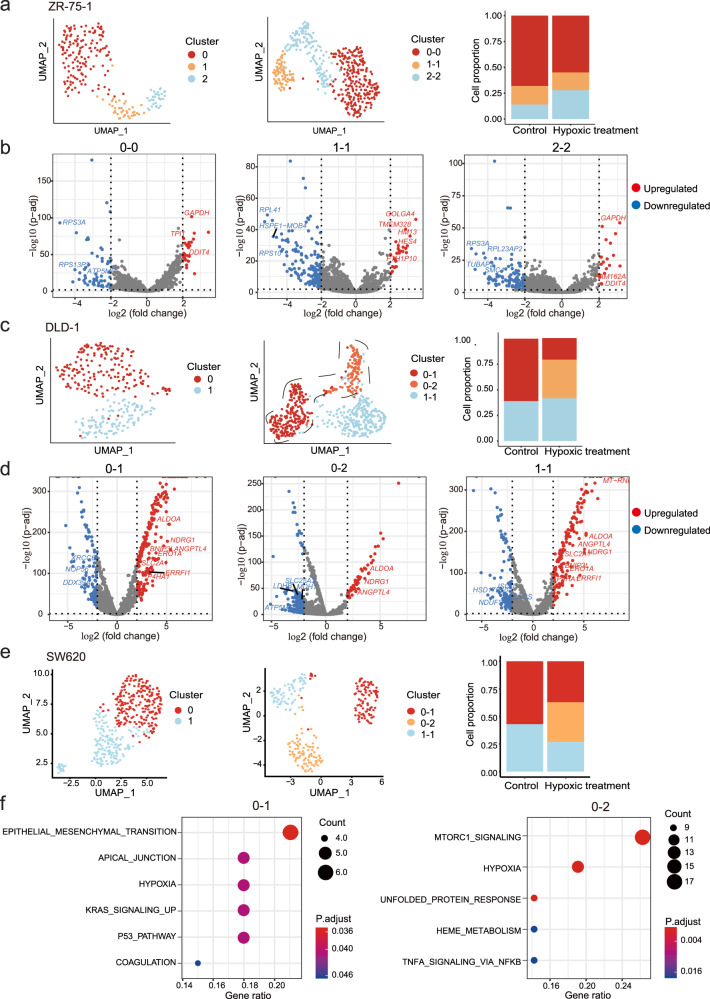
Human cancer cell lines have long served as tools for cancer research and drug discovery, but the presence and the source of intra-cell-line heterogeneity remain elusive. Here, we perform single-cell RNA-sequencing and ATAC-sequencing on 42 and 39 human cell lines, respectively, to illustrate both transcriptomic and epigenetic heterogeneity within individual cell lines. Our data reveal that transcriptomic heterogeneity is frequently observed in cancer cell lines of different tissue origins, often driven by multiple common transcriptional programs. Copy number variation, as well as epigenetic variation and extrachromosomal DNA distribution all contribute to the detected intra-cell-line heterogeneity. Using hypoxia treatment as an example, we demonstrate that transcriptomic heterogeneity could be reshaped by environmental stress. Overall, our study performs single-cell multi-omics of commonly used human cancer cell lines and offers mechanistic insights into the intra-cell-line heterogeneity and its dynamics, which would serve as an important resource for future cancer cell line-based studies.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Method of the Year 2019: single-cell multimodal omics. Nat. Methods17, 1 (2020). - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
- 2021YFF1201000 and 2022YFC3400400/Chinese Ministry of Science and Technology | Department of S and T for Social Development (Department of S&T for Social Development)
- KQTD20180411143432337/Shenzhen Science and Technology Innovation Commission
- ZDSYS20200811144002008/Shenzhen Science and Technology Innovation Commission
- JCYJ20210324104605014/Shenzhen Science and Technology Innovation Commission
- ZDSYS20190902093613831/Shenzhen Science and Technology Innovation Commission
LinkOut - more resources
Full Text Sources
Medical
Research Materials
