

Distinct Hodgkin lymphoma subtypes defined by noninvasive genomic profiling
- PMID: 38081297
- PMCID: PMC11293530
- DOI: 10.1038/s41586-023-06903-x
Distinct Hodgkin lymphoma subtypes defined by noninvasive genomic profiling
Abstract
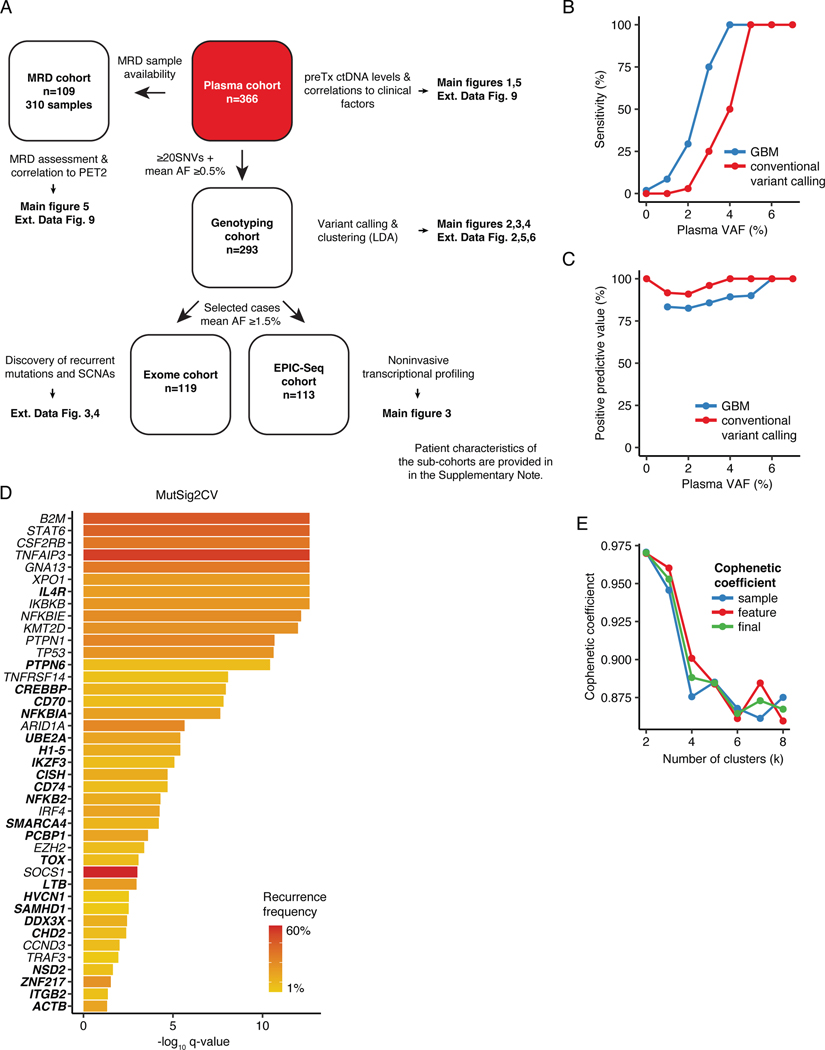
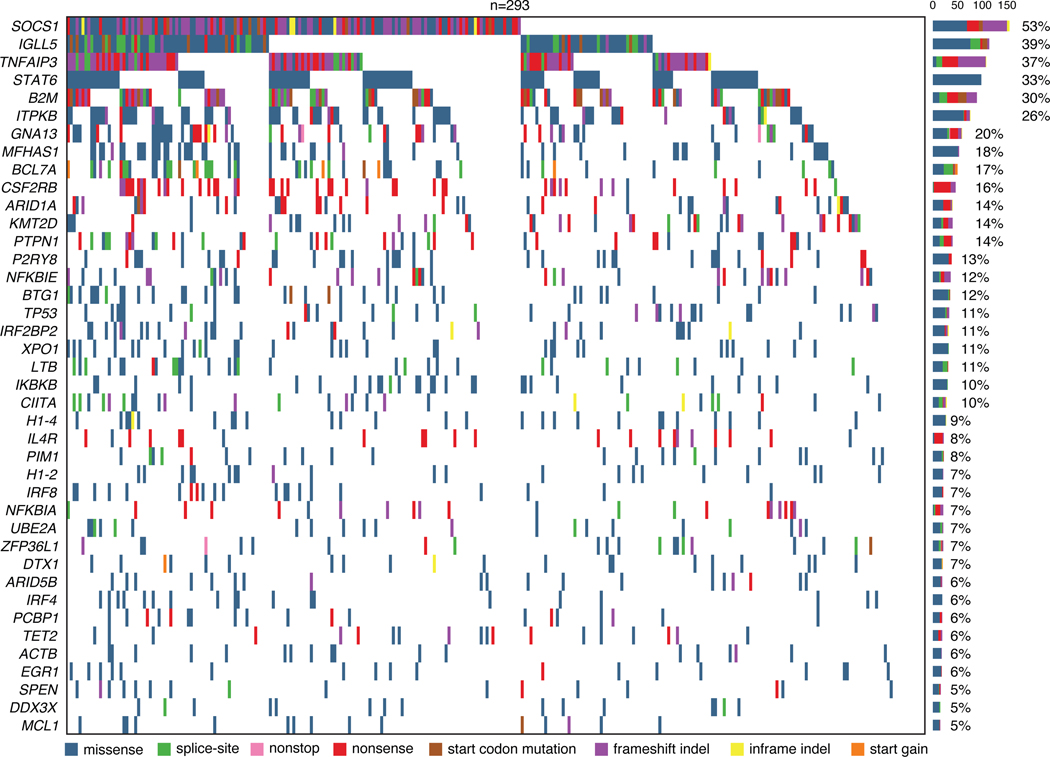
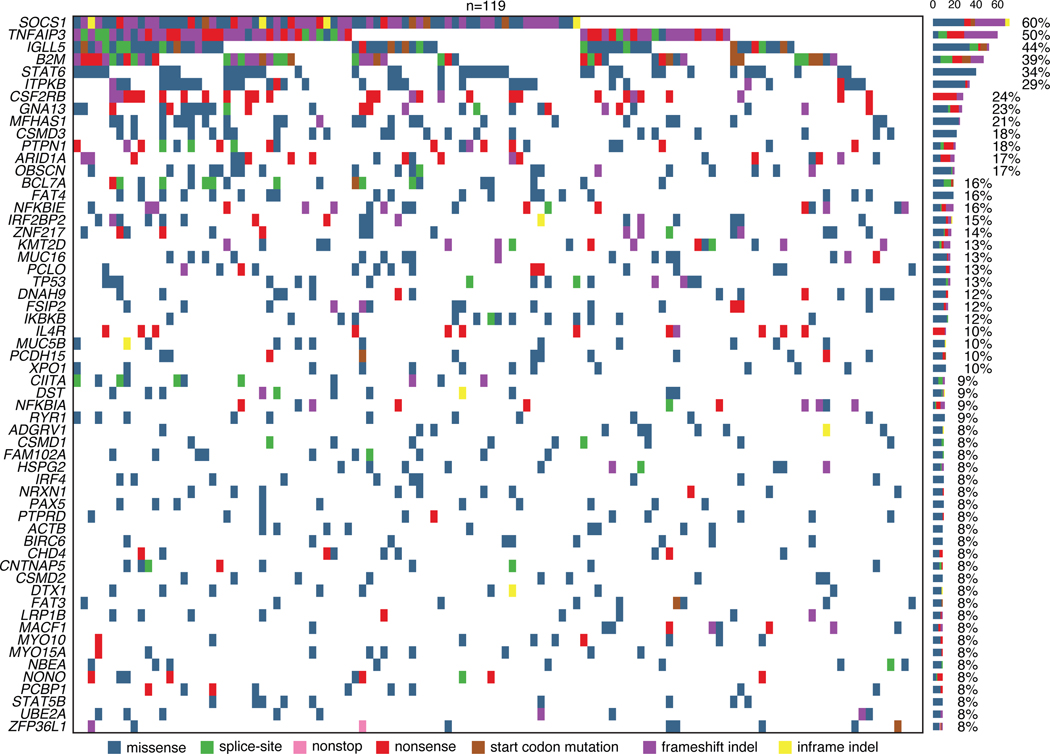
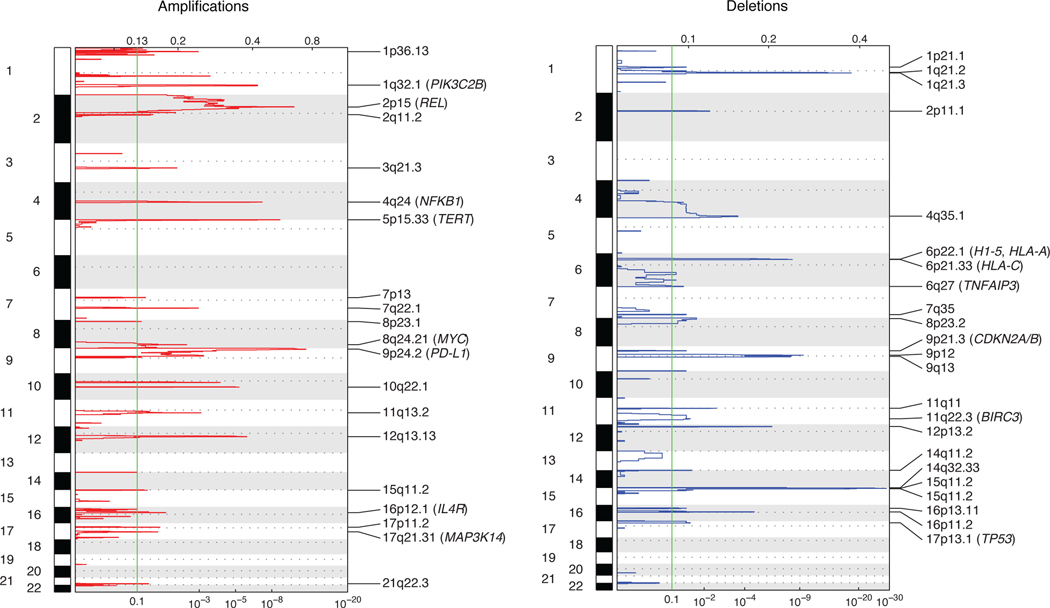
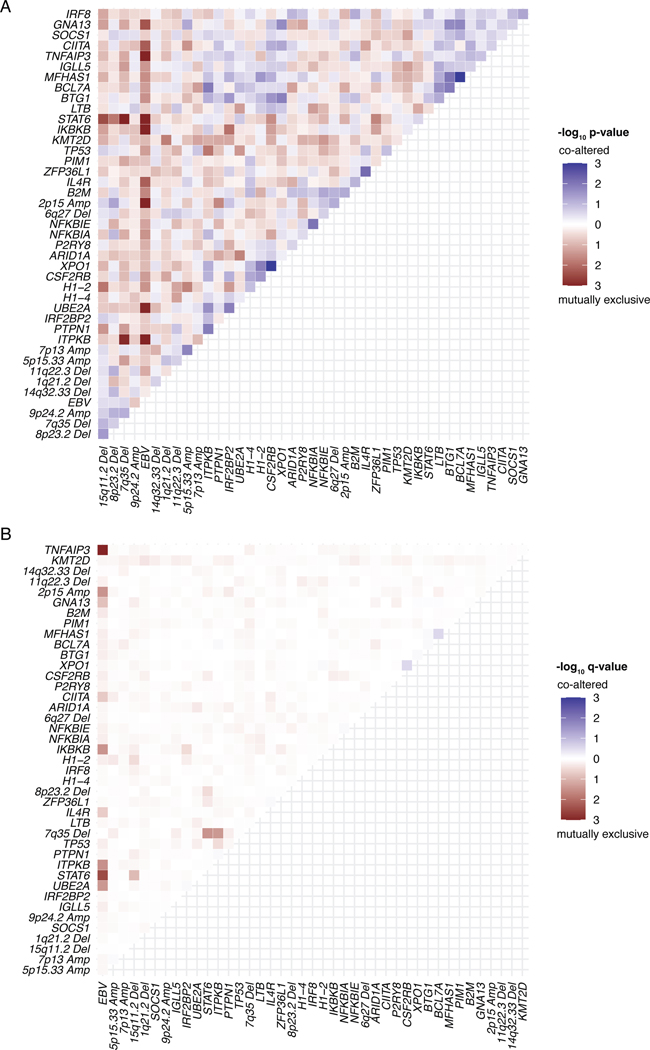
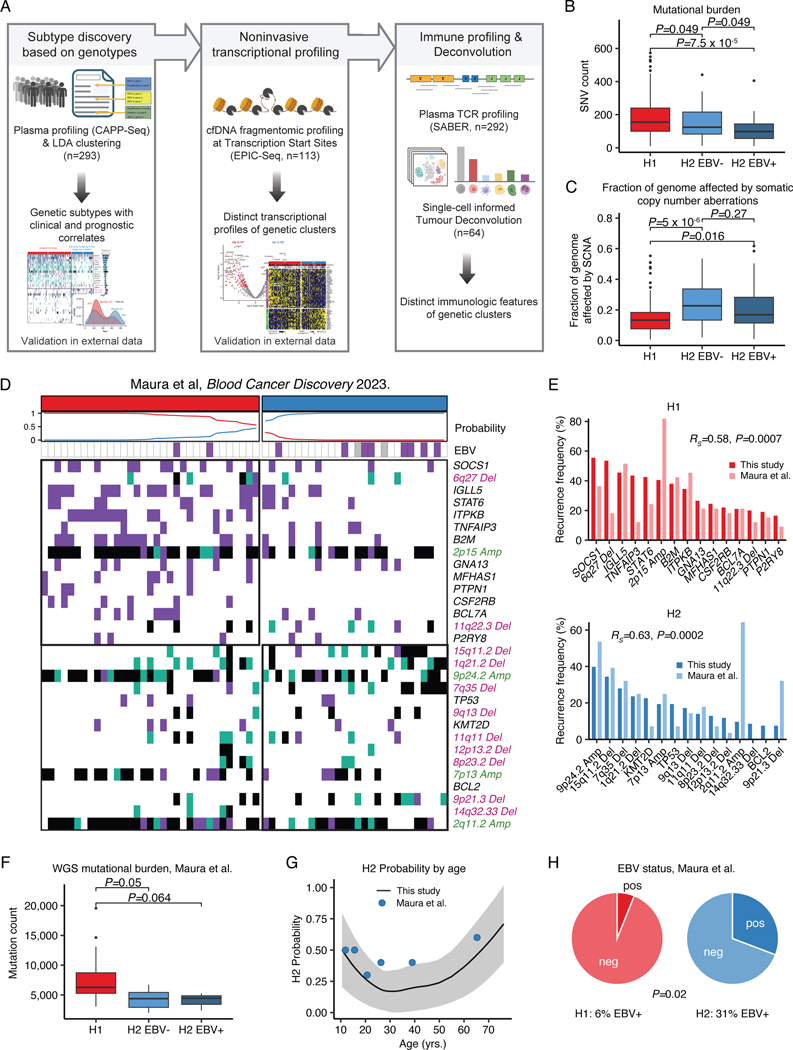
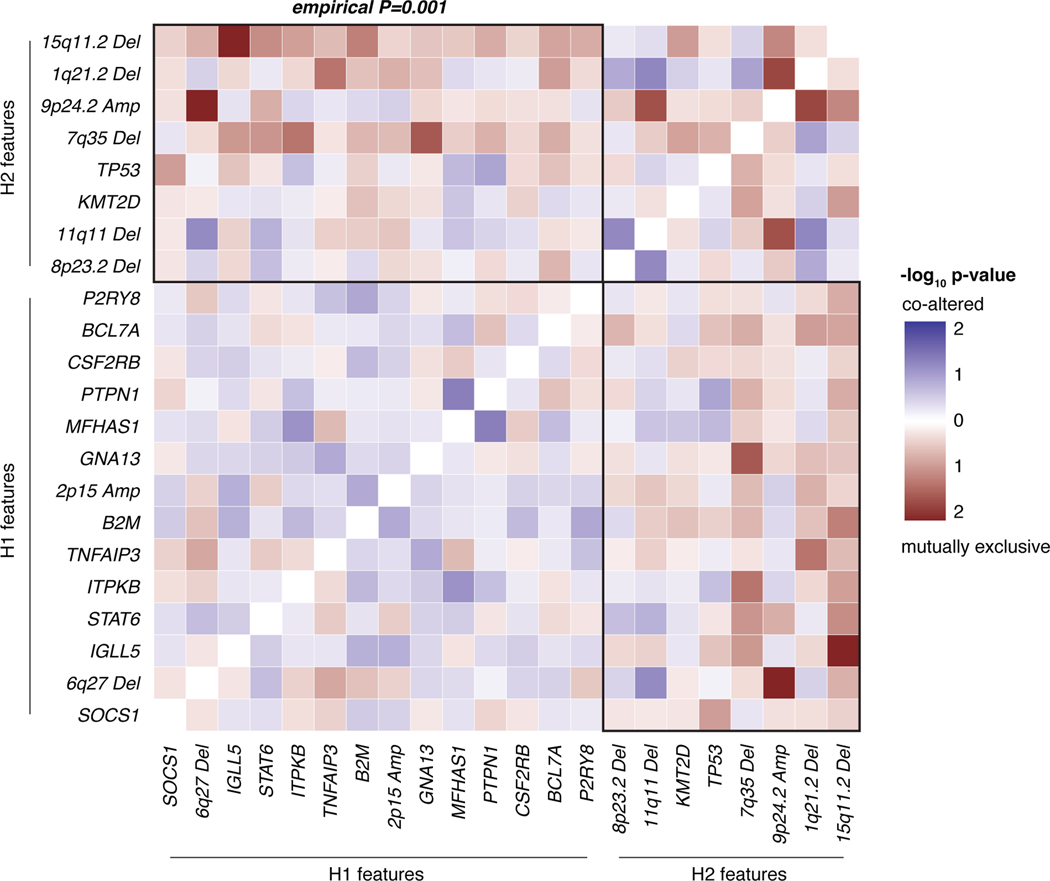
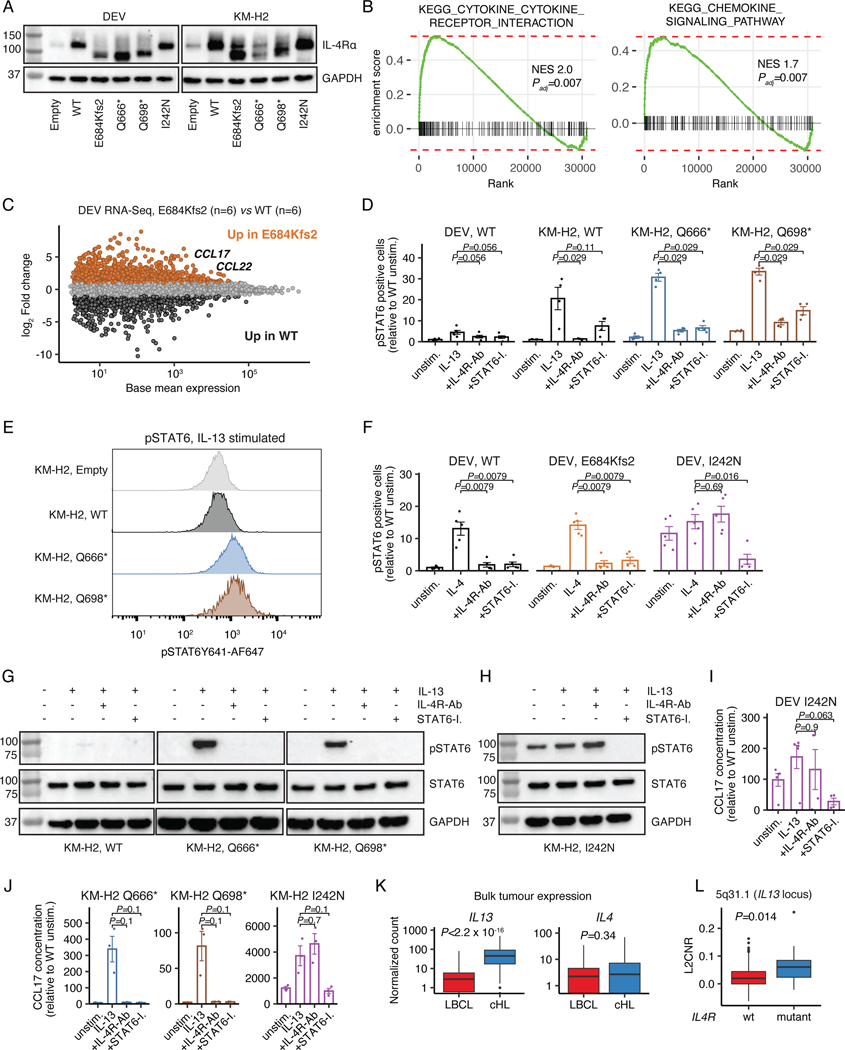
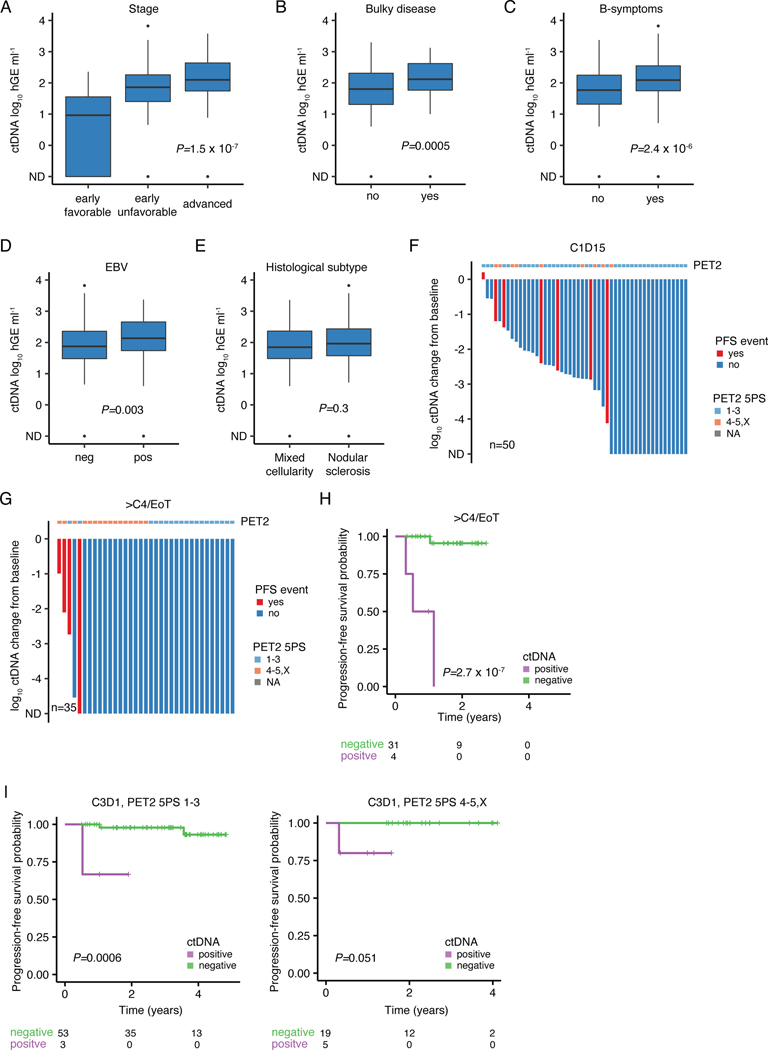
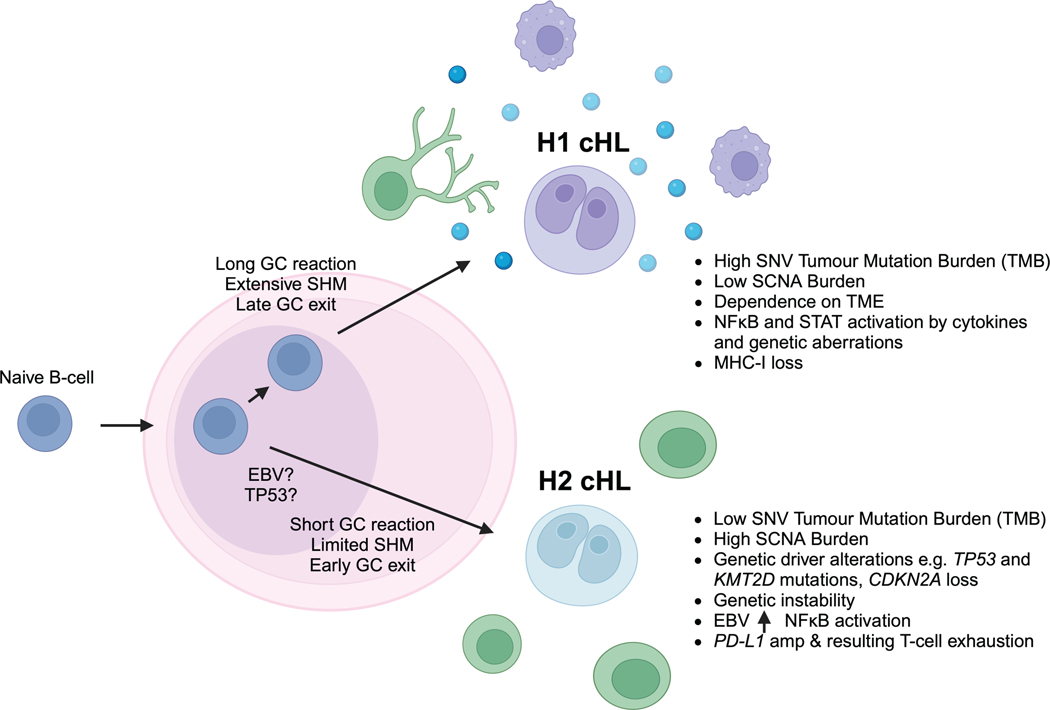
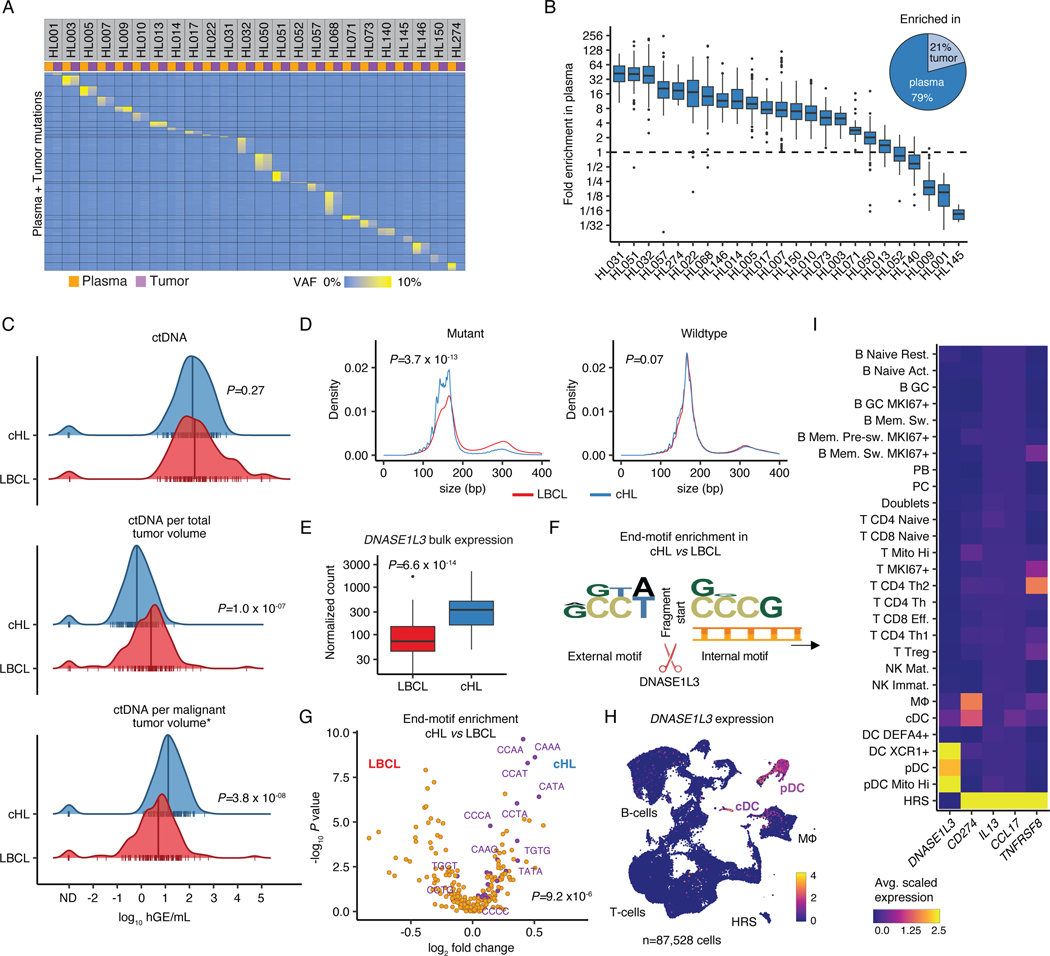
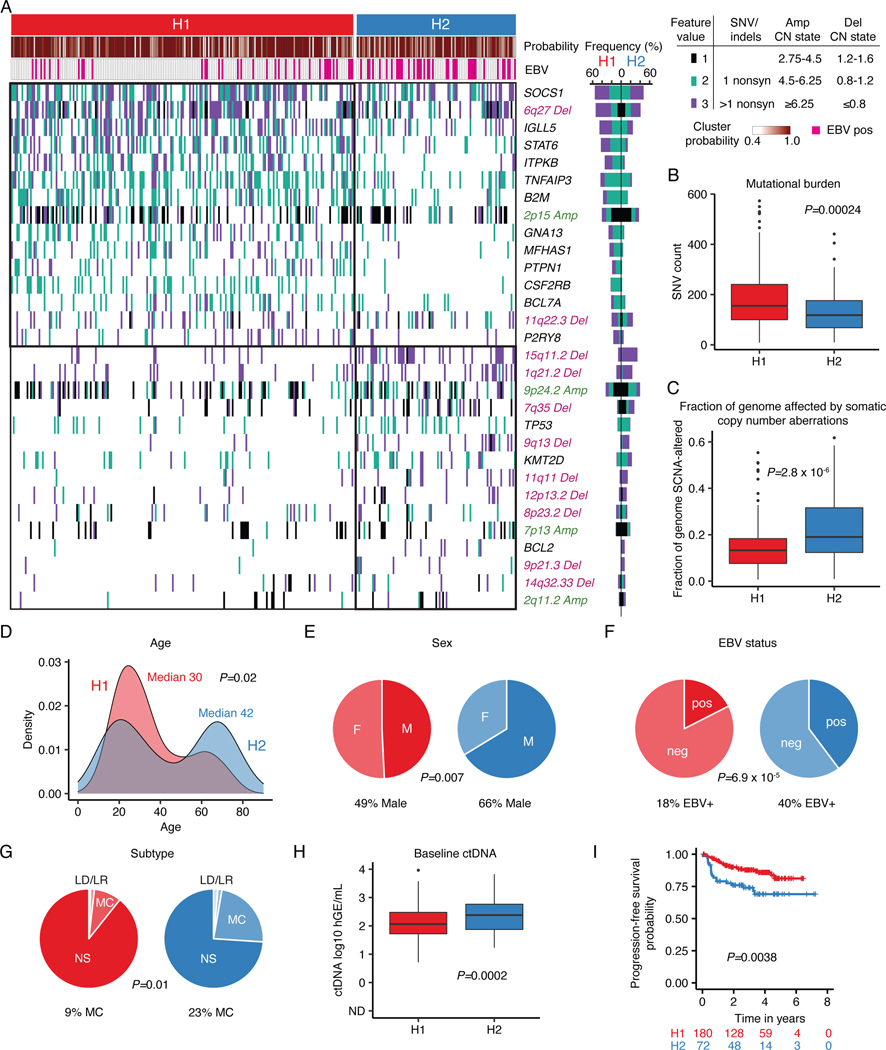
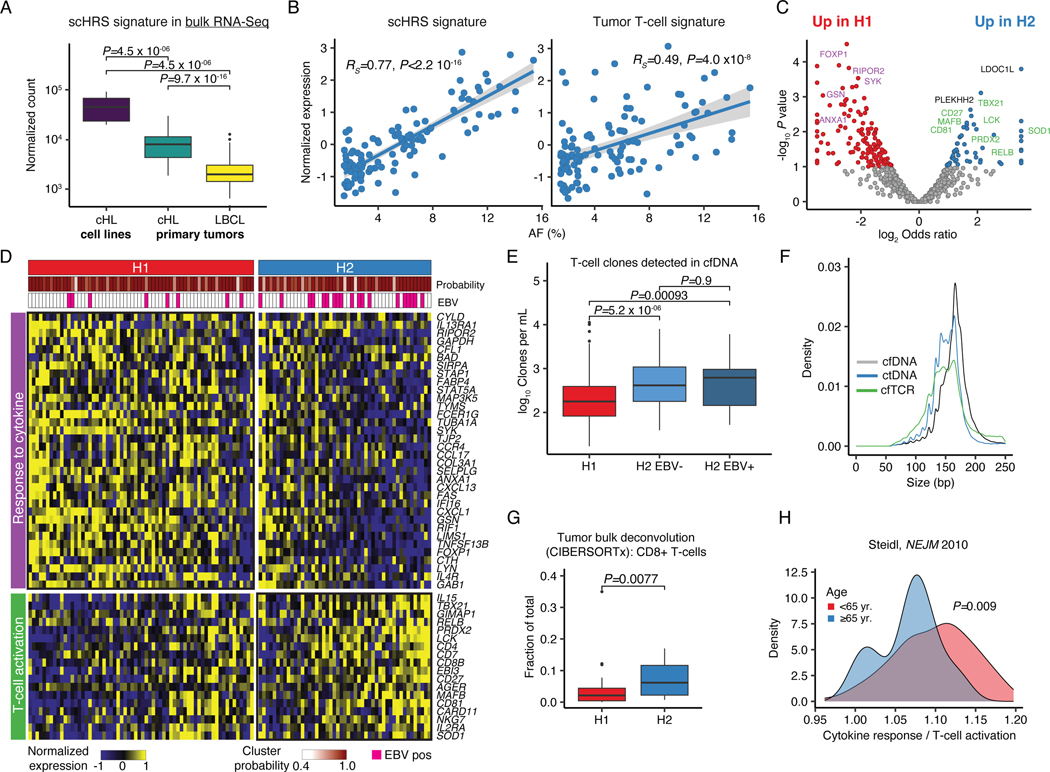
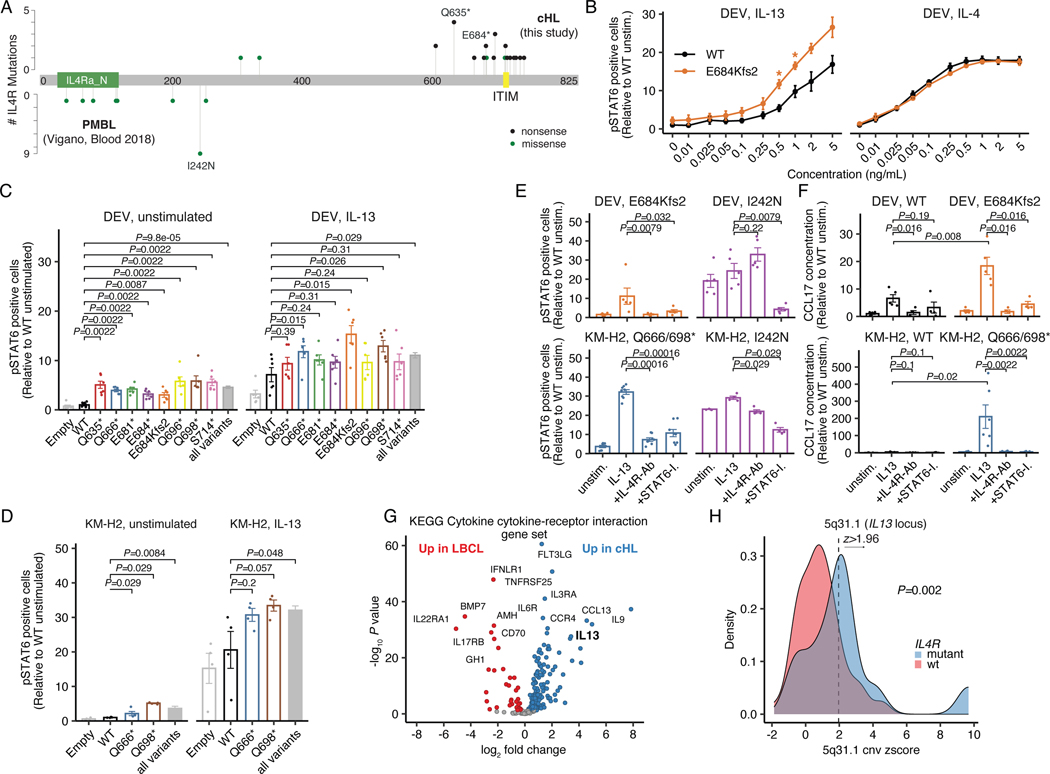
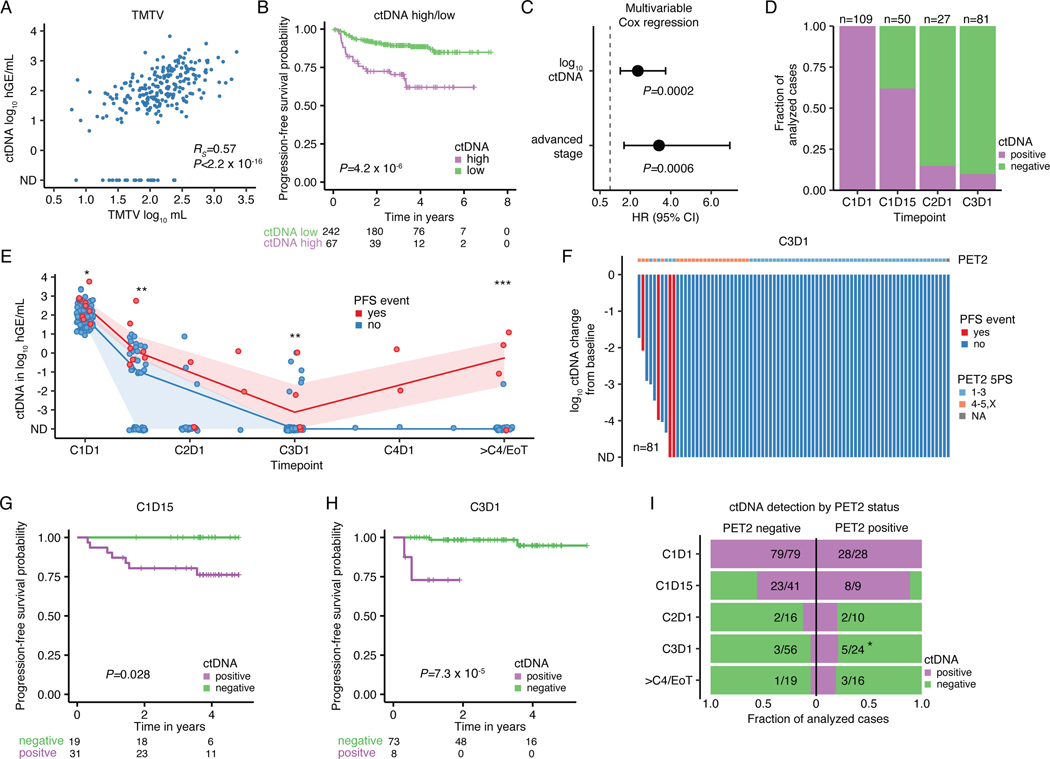
The scarcity of malignant Hodgkin and Reed-Sternberg cells hampers tissue-based comprehensive genomic profiling of classic Hodgkin lymphoma (cHL). By contrast, liquid biopsies show promise for molecular profiling of cHL due to relatively high circulating tumour DNA (ctDNA) levels1-4. Here we show that the plasma representation of mutations exceeds the bulk tumour representation in most cases, making cHL particularly amenable to noninvasive profiling. Leveraging single-cell transcriptional profiles of cHL tumours, we demonstrate Hodgkin and Reed-Sternberg ctDNA shedding to be shaped by DNASE1L3, whose increased tumour microenvironment-derived expression drives high ctDNA concentrations. Using this insight, we comprehensively profile 366 patients, revealing two distinct cHL genomic subtypes with characteristic clinical and prognostic correlates, as well as distinct transcriptional and immunological profiles. Furthermore, we identify a novel class of truncating IL4R mutations that are dependent on IL-13 signalling and therapeutically targetable with IL-4Rα-blocking antibodies. Finally, using PhasED-seq5, we demonstrate the clinical value of pretreatment and on-treatment ctDNA levels for longitudinally refining cHL risk prediction and for detection of radiographically occult minimal residual disease. Collectively, these results support the utility of noninvasive strategies for genotyping and dynamic monitoring of cHL, as well as capturing molecularly distinct subtypes with diagnostic, prognostic and therapeutic potential.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
Figures
References
-
- Vandenberghe P et al. Non-invasive detection of genomic imbalances in Hodgkin/Reed-Sternberg cells in early and advanced stage Hodgkin’s lymphoma by sequencing of circulating cell-free DNA: a technical proof-of-principle study. The Lancet Haematology 2, e55–e65 (2015). 10.1016/s2352-3026(14)00039-8 - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
