

Assessing computational tools for predicting protein stability changes upon missense mutations using a new dataset
- PMID: 38084013
- PMCID: PMC10751734
- DOI: 10.1002/pro.4861
Assessing computational tools for predicting protein stability changes upon missense mutations using a new dataset
Abstract
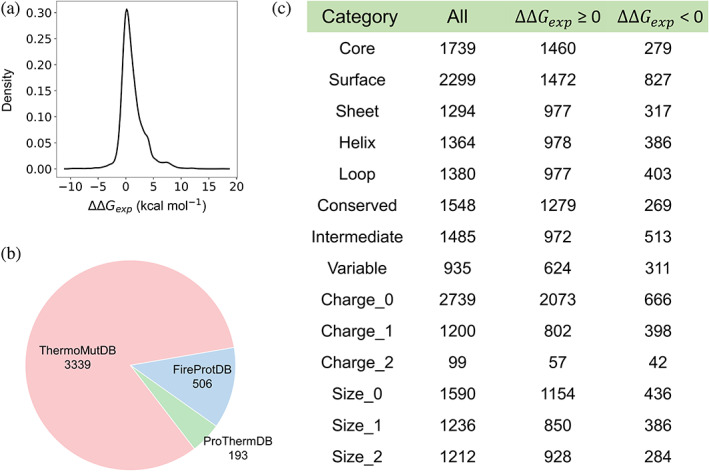
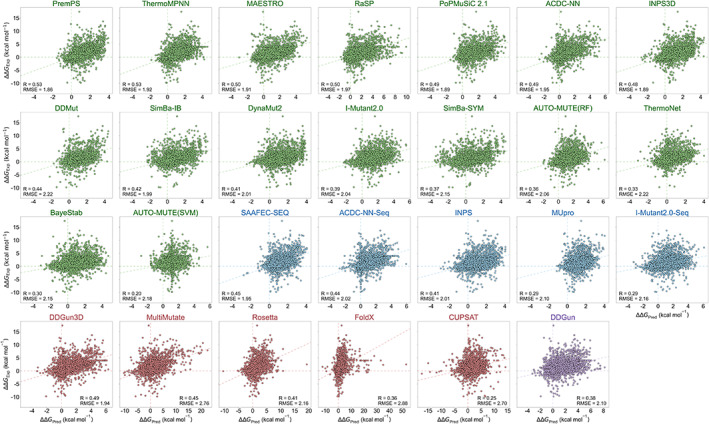
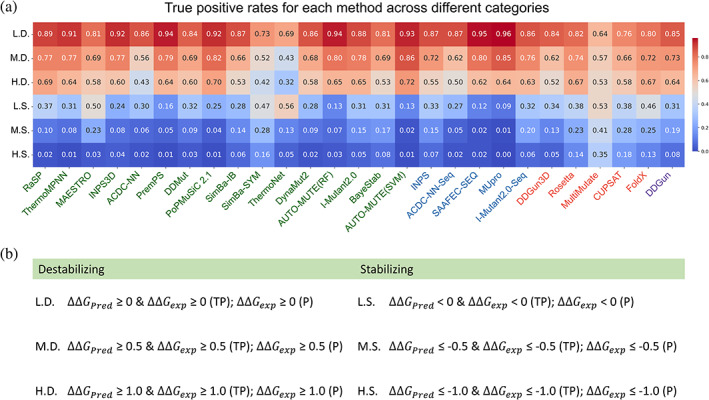
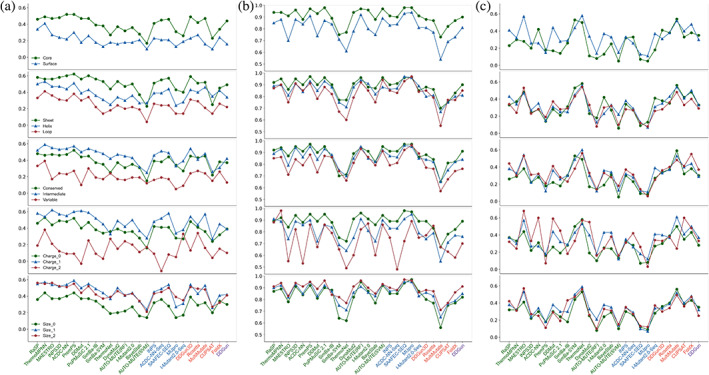
Insight into how mutations affect protein stability is crucial for protein engineering, understanding genetic diseases, and exploring protein evolution. Numerous computational methods have been developed to predict the impact of amino acid substitutions on protein stability. Nevertheless, comparing these methods poses challenges due to variations in their training data. Moreover, it is observed that they tend to perform better at predicting destabilizing mutations than stabilizing ones. Here, we meticulously compiled a new dataset from three recently published databases: ThermoMutDB, FireProtDB, and ProThermDB. This dataset, which does not overlap with the well-established S2648 dataset, consists of 4038 single-point mutations, including over 1000 stabilizing mutations. We assessed these mutations using 27 computational methods, including the latest ones utilizing mega-scale stability datasets and transfer learning. We excluded entries with overlap or similarity to training datasets to ensure fairness. Pearson correlation coefficients for the tested tools ranged from 0.20 to 0.53 on unseen data, and none of the methods could accurately predict stabilizing mutations, even those performing well in anti-symmetric property analysis. While most methods present consistent trends for predicting destabilizing mutations across various properties such as solvent exposure and secondary conformation, stabilizing mutations do not exhibit a clear pattern. Our study also suggests that solely addressing training dataset bias may not significantly enhance accuracy of predicting stabilizing mutations. These findings emphasize the importance of developing precise predictive methods for stabilizing mutations.
Keywords: computational tools; missense mutations; protein stability changes; stabilizing mutations.
© 2023 The Protein Society.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Assessing the performance of computational predictors for estimating protein stability changes upon missense mutations.Brief Bioinform. 2021 Nov 5;22(6):bbab184. doi: 10.1093/bib/bbab184. Brief Bioinform. 2021. PMID: 34058752
-
PremPS: Predicting the impact of missense mutations on protein stability.PLoS Comput Biol. 2020 Dec 30;16(12):e1008543. doi: 10.1371/journal.pcbi.1008543. eCollection 2020 Dec. PLoS Comput Biol. 2020. PMID: 33378330 Free PMC article.
-
Predicting protein stability changes upon single-point mutation: a thorough comparison of the available tools on a new dataset.Brief Bioinform. 2022 Mar 10;23(2):bbab555. doi: 10.1093/bib/bbab555. Brief Bioinform. 2022. PMID: 35021190 Free PMC article.
-
Reviewing Challenges of Predicting Protein Melting Temperature Change Upon Mutation Through the Full Analysis of a Highly Detailed Dataset with High-Resolution Structures.Mol Biotechnol. 2021 Oct;63(10):863-884. doi: 10.1007/s12033-021-00349-0. Epub 2021 Jun 8. Mol Biotechnol. 2021. PMID: 34101125 Free PMC article. Review.
-
Predicting the stability of mutant proteins by computational approaches: an overview.Brief Bioinform. 2021 May 20;22(3):bbaa074. doi: 10.1093/bib/bbaa074. Brief Bioinform. 2021. PMID: 32496523 Review.
Cited by
-
Exploring Evolution to Uncover Insights Into Protein Mutational Stability.Mol Biol Evol. 2025 Jan 6;42(1):msae267. doi: 10.1093/molbev/msae267. Mol Biol Evol. 2025. PMID: 39786559 Free PMC article.
-
Leveraging computer-aided design and artificial intelligence to develop a next-generation multi-epitope tuberculosis vaccine candidate.Infect Med (Beijing). 2024 Nov 9;3(4):100148. doi: 10.1016/j.imj.2024.100148. eCollection 2024 Dec. Infect Med (Beijing). 2024. PMID: 39687693 Free PMC article.
-
Revolutionizing Molecular Design for Innovative Therapeutic Applications through Artificial Intelligence.Molecules. 2024 Sep 29;29(19):4626. doi: 10.3390/molecules29194626. Molecules. 2024. PMID: 39407556 Free PMC article. Review.
-
Transfer Learning in Cancer Genetics, Mutation Detection, Gene Expression Analysis, and Syndrome Recognition.Cancers (Basel). 2024 Jun 4;16(11):2138. doi: 10.3390/cancers16112138. Cancers (Basel). 2024. PMID: 38893257 Free PMC article. Review.
-
The origin of mutational epistasis.Eur Biophys J. 2024 Nov;53(7-8):473-480. doi: 10.1007/s00249-024-01725-9. Epub 2024 Oct 23. Eur Biophys J. 2024. PMID: 39443382
References
-
- Baek KT, Kepp KP. Data set and fitting dependencies when estimating protein mutant stability: toward simple, balanced, and interpretable models. J Comput Chem. 2022;43:504–518. - PubMed
-
- Benevenuta S, Pancotti C, Fariselli P, Birolo G, Sanavia T. An antisymmetric neural network to predict free energy changes in protein variants. J Phys D Appl Phys. 2021;54:245403.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
