

miR-222 inhibits pathological cardiac hypertrophy and heart failure
- PMID: 38084908
- PMCID: PMC10939454
- DOI: 10.1093/cvr/cvad184
miR-222 inhibits pathological cardiac hypertrophy and heart failure
Abstract
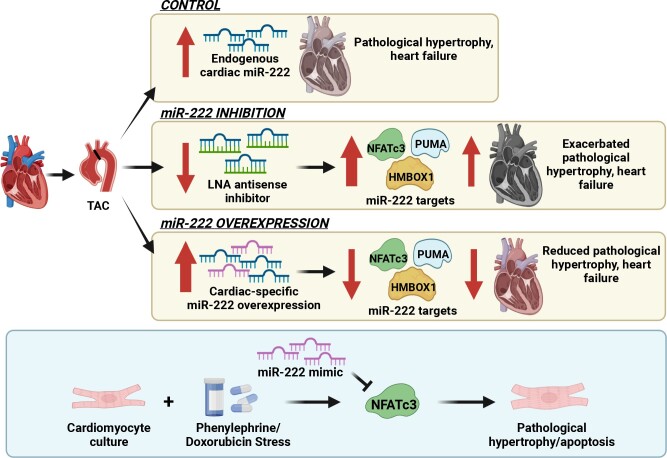
Aims: Physiological cardiac hypertrophy occurs in response to exercise and can protect against pathological stress. In contrast, pathological hypertrophy occurs in disease and often precedes heart failure. The cardiac pathways activated in physiological and pathological hypertrophy are largely distinct. Our prior work demonstrated that miR-222 increases in exercised hearts and is required for exercise-induced cardiac hypertrophy and cardiomyogenesis. Here, we sought to define the role of miR-222 in pathological hypertrophy.
Methods and results: We found that miR-222 also increased in pathological hypertrophy induced by pressure overload. To assess its functional significance in this setting, we generated a miR-222 gain-of-function model through cardiac-specific constitutive transgenic miR-222 expression (TgC-miR-222) and used locked nucleic acid anti-miR specific for miR-222 to inhibit its effects. Both gain- and loss-of-function models manifested normal cardiac structure and function at baseline. However, after transverse aortic constriction (TAC), miR-222 inhibition accelerated the development of pathological hypertrophy, cardiac dysfunction, and heart failure. Conversely, miR-222-overexpressing mice had less pathological hypertrophy after TAC, as well as better cardiac function and survival. We identified p53-up-regulated modulator of apoptosis, a pro-apoptotic Bcl-2 family member, and the transcription factors, Hmbox1 and nuclear factor of activated T-cells 3, as direct miR-222 targets contributing to its roles in this context.
Conclusion: While miR-222 is necessary for physiological cardiac growth, it inhibits cardiac growth in response to pressure overload and reduces adverse remodelling and cardiac dysfunction. These findings support the model that physiological and pathological hypertrophy are fundamentally different. Further, they suggest that miR-222 may hold promise as a therapeutic target in pathological cardiac hypertrophy and heart failure.
Keywords: Heart failure; MicroRNA; Pathological hypertrophy; Transverse aortic constriction (TAC).
© The Author(s) 2023. Published by Oxford University Press on behalf of the European Society of Cardiology. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Conflict of interest statement
Conflict of interest: none declared.
Figures




Comment in
-
The dual effects of miR-222 in cardiac hypertrophy: bridging pathological and physiological paradigms.Cardiovasc Res. 2024 Mar 14;120(3):217-219. doi: 10.1093/cvr/cvae033. Cardiovasc Res. 2024. PMID: 38484215 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
